
Je ne suis pas biologiste. J'ai simplement fait de la biologie
pendant mes études à l'X (c'était une de mes majeures). J'ai
continué ensuite à m'y intéresser pendant les années qui ont suivi. En
2011, je me suis retrouvé impliqué dans un projet relativement
important de bio-informatique, ce qui m'a amené à me replonger
sérieusement sur le sujet. Je me suis alors aperçu que
l'information nécessaire à un informaticien pour comprendre les
bases qui lui sont nécessaires dans ce genre d'activité existe
bien entendu, mais est relativement dispersée, ou diluée à
l'intérieur de sites internets ou de différents ouvrages, parfois
techniques et peu accessibles à quelqu'un qui n'a pas déjà de
solides bases en biologie. C'est ce qui m'a amené à rédiger cette
page.
Le but de cette page n'étant pas de faire un cours de génétique, mais
simplement de présenter des éléments fondamentaux, elle se limite
globalement à la présentation des cellules somatiques (qui ne sont
pas à l'origine des gamètes), eucaryotes (avec un noyau) et
diploïdes (avec des chromosomes présents par paire). Les exemples
seront pris sur l'homme et le maïs
N'étant pas spécialiste, il est tout à fait possible qu'elle contienne
des erreurs ou des omissions importantes: elle ne reflète que ce que j'ai
compris. Il ne faut donc certainement pas prendre ce texte pour
plus que ce qu'il est et se garder de le considérer comme une
référence. Il s'agit avant tout de vulgarisation. Si un biologiste
"professionnel" venait à la lire, ses
remarques seraient bienvenues!
La majorité des illustrations ont été prises sur wikimedia (sauf mention contraire), et parfois retravaillées.
Une cellule eucaryote est composé de plusieurs entités, mais deux seulement d'entre elles contiennent un code génétique contenu dans de l'ADN (acide désoxyribonucléique), les mitochondries et le noyau.
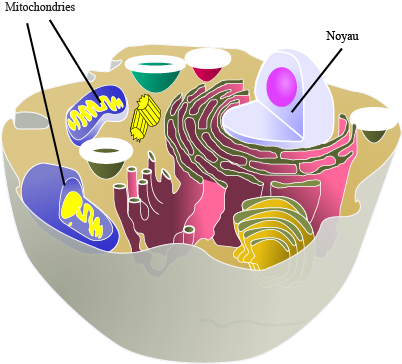
Les mitochondries sont les "piles à combustible" de la cellule. Elles
disposent de leur propre ADN (ADN mitochondrial), qui est transmis
exclusivement par la mère. C'est ainsi que l'on peut tracer les
lignées maternelles, comme l'on peut utiliser le chromosome Y chez
l'homme pour tracer les lignées paternelles.
Lorsque
l'on parle de l'ADN sans autre précision, il s'agit de l'ADN contenu dans le
noyau de la cellule. L'ADN présent dans le noyau de la
cellule se trouve sous forme de chromatine, une combinaison de l'ADN avec
des protéines qui en assurent "l'emballage". La forme de l'ADN varie
en fonction du cycle cellulaire, mais il ne prend sa forme
caractéristique de chromosomes qu'au moment de la division cellulaire
(la mitose).
Le reste du temps il apparaît sous forme de filaments
relativement indifférenciés, qui se regrouperont donc en chromosomes
au moment de la division cellulaire.
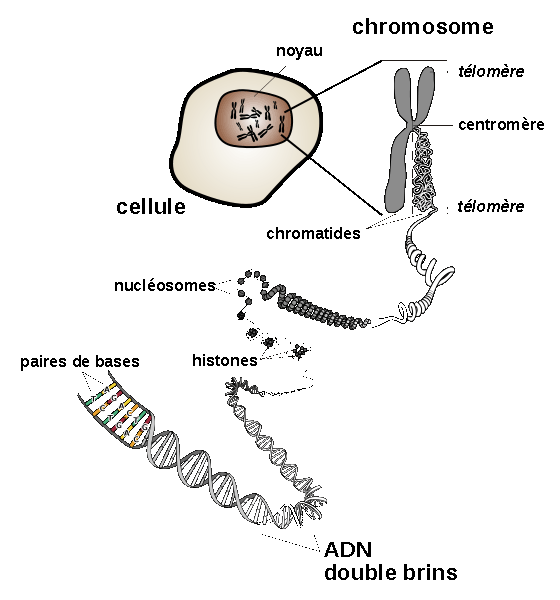
La forme d'un chromosome juste avant la division cellulaire est
généralement celle d'un X avec 4 bras
appelés chromatides, reliés en leur centre par le centromère (on parle
de chromosomes métacentriques si le centromère est approximativement au
milieu, d'acrocentriques s'il est proche d'une extrémité, et de
télocentriques si le centromère est à une extrémité et que le
chromosome n'a qu'un bras). L'extrémité des bras s'appelle
le télomère.
Les télomères ont une grande importance pour
comprendre le vieillissement, car ils raccourcissent à chaque division
cellulaire, ce qui peut entraîner à terme une sénescence et une
apoptose de la cellule (mort cellulaire). Une enzyme, la télomérase,
permet de faire croître les télomères, mais sa synthèse est
généralement inhibée chez l'être humain sauf dans les cellules
indifférenciées (cellules souches). La télomérase est soupçonnée
d'être nécessaire à la reproduction des cellules cancéreuses, qui se
divisent indéfiniment. Le rôle exact du raccourcissement des
télomères est encore mal compris..
Les bras courts sont appelés bras p (pour petit) et les bras longs sont les bras q. Les deux bras courts (resp. les deux bras longs) sont strictement identiques pour un même chromosome, et sont appelés chromatides soeurs. Ces chromatides soeurs existent en raison de la duplication de l'ADN qui a eu lieu juste avant la phase de regroupement des chromosomes. Lors de la division de la cellule, les chromosomes vont se partager en deux et le matériel génétique se retrouvera à nouveau en un seul exemplaire dans chacune des cellules.
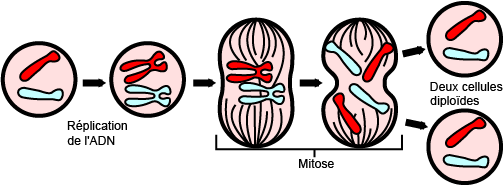
Étapes de la division cellulaire
L'ADN n'existe sous forme de chromosome qu'à un stade très
particulier de la vie de la cellule.
Tous les individus diploïdes (c'est notre cas) possèdent deux chromosomes de chaque type (appelés chromosomes autosomes), à l'exception des chromosomes sexuels qui forment aussi une paire (ils sont alors appelés chromosomes homologues), mais qui ne sont de même type que chez la femme (2 chromosomes X), alors qu'ils sont différents chez l'homme (un X et un Y). L'être humain possède 22 paires de chromosomes autosomes (numérotés de 1 à 22), et une paire de chromosomes sexuels homologues, soit 46 chromosomes par cellule.
Il faut cependant bien
comprendre que si nous avons deux chromosomes 1, deux chromosomes 2,
etc, ces deux chromosomes autosomes, de même type et visuellement identiques
sont cependant différents:
l'ADN qu'ils contiennent n'est pas
exactement le même. L'un nous a été légué par notre père et l'autre
par notre mère. Ils contiennent les gènes permettant de coder les
mêmes fonctions (comme par exemple la fabrication de la mélanine),
mais de façon subtilement différente (comme par exemple la quantité de
mélanine produite). Les différentes variations d'un même gène sont
appelés allèles.
Une erreur classique chez nombre de débutants en biologie est de se
"mélanger les pinceaux" entre les chromatides soeurs sur un chromosome
(qui sont strictement identiques et portent exactement le même code
génétique puisque issus de la réplication de l'ADN dans la cellule
juste avant la mitose), et les deux chromosomes autosomes
qui sont visuellement identiques mais pourtant bien différents.
Attention à la confusion...
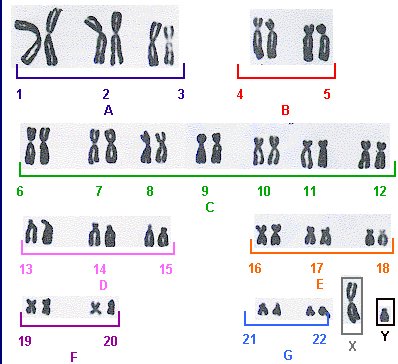
Caryotype humain
Comme on peut le voir sur un caryotype, les chromosomes humains sont très
différents en taille et en structure. Les chromosomes 1 et 2 sont très
grands, les chromosomes 21, 22 ou Y sont beaucoup plus petits. Un
dernier (mais important) rappel: la représentation de l'ADN sous forme
de chromosomes
est faite à un stade très particulier de la vie de la cellule, juste
après la duplication de l'ADN. Il y a donc deux fois plus d'ADN (c'est
la raison de l'existence des chromatides soeurs) dans
la cellule à ce moment là qu'en "temps normal" de la vie
cellulaire.
Chez la femme, qui possède deux chromosomes X, l'un des deux est
inactivé et n'exprimera jamais son code génétique. Donc un être
humain, de quelque sexe qu'il soit, ne possède qu'un seul chromosome X
actif. Le fait de posséder un chromosome en plus (trisomie), ou en
moins (monosomie) est généralement létal, surtout pour les grands
chromosomes qui codent de nombreuses fonctions. Pour les petits
chromosomes, il existe des trisomies non systématiquement fatales
comme la trisomie 21 (vulgairement appelé "mongolisme", 1 embryon sur
7 survit), ou la trisomie 22 partielle (appelé "maladie des yeux de
chat", une partie seulement du chromosome 22 est dupliqué). Les
trisomies 13 et
18 permettent parfois d'amener les grossesses à terme mais la majorité
des enfants décèdent avant l'âge d'un an. Les trisomies des
chromosomes sexuels ne sont pas létales et même parfois
non détectés avant l'âge adulte (XXY -> syndrome
de Klinefelter -> individu mâle stérile, XXX -> syndrome
triple X, individu de sexe féminin, parfois stérile).
La méiose est la division cellulaire qui permet la reproduction sexuée. Partant d'une cellule à 2n chromosomes (nombre normal de chromosomes), elle aboutit à 4 cellules ne contenant que n chromosomes, soit 4 gamètes (ovules chez la femme, spermatozoïde chez l'homme).
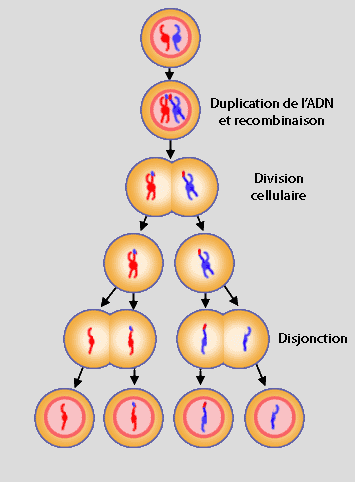
Schéma général de la méiose
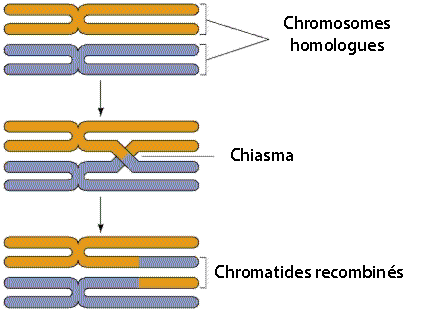
Exemple de recombinaison
élémentaire des chromatides
Arrivé à ce stade, les gamètes sont achevés. La fertilisation d'un gamète femelle par un gamète mâle combinera une cellule à n chromosomes avec une autre cellule à n chromosome pour aboutir à nouveau à une cellule "classique" à 2n chromosomes.
Il faut noter que les chromosomes X et Y ne peuvent pas se combiner,
car ils sont structurellement différents. Chez les hommes (qui
possèdent un X et un Y), le
chromosome Y est passé "à l'identique" de père en fils. En revanche,
chez les femmes, les deux chromosomes X se recombinent comme les
chromosomes autosomes, et le X transmis est la combinaison du X de la
grand-mère et du grand-père maternel. Le fils, ou la fille, possède
donc toujours l'X recombiné provenant de leur mère (recombinaison des
chromosomes X de ses grands-parents maternels). En ce qui concerne le
second chromosome sexuel transmis par le père, un
garçon possédera un Y venu de sa lignée paternelle sans aucune
modification, alors que la fille aura un second X transmis à son père
par sa grand-mère paternelle (et qui est la recombinaison des
chromosomes X transmis à sa grand-mère par les deux arrières
grands-parents). Ouf...
Le fait que le chromosome Y soit transmis sans recombinaison
aboutit à ce que l'on appelle la dégénérescence du chromosome
Y. Au fil de l'évolution, les mutations se sont accumulées dans le
chromosome Y, sans espoir de réparation au travers des recombinaisons
de la reproduction sexuée, aboutissant à l'inactivation de nombre de
ses gènes.
Un évènement central de la vie cellulaire est sa capacité à se dupliquer, et pour ce faire à dupliquer exactement, sans erreur de copie, son ADN. Le mécanisme de copie de l'ADN a été élucidé quand James Watson et Francis Crick ont établi la structure en double hélice de l'ADN entre 1951 et 1953.
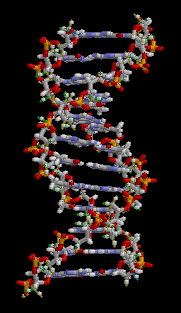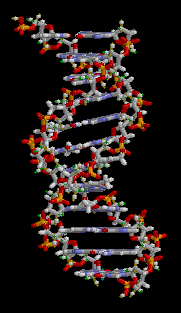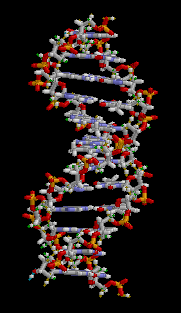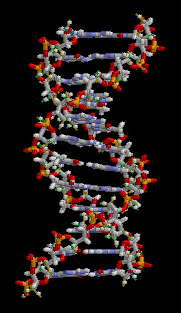
Structure 3D de l'ADN
L'ADN est composé de deux longs brins complémentaires de polymères composés d'unités simples appelés nucléotides.
Structure 3D de l'ADN
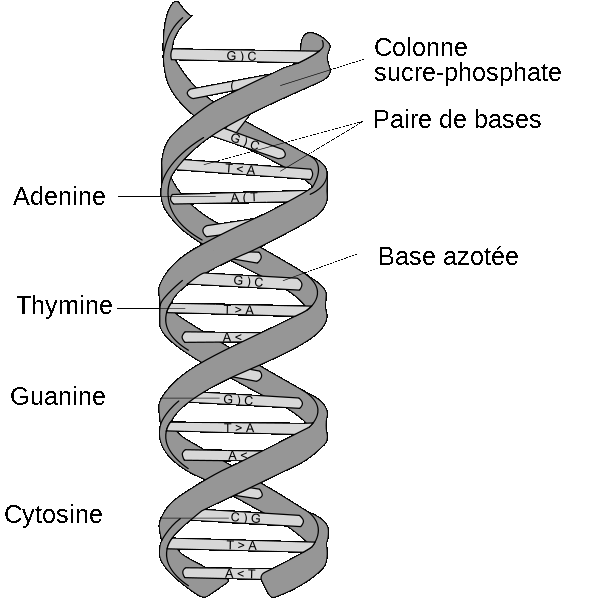
Les nucléotides sont composés de bases azotées, de sucres (les desoxyriboses) et de phosphate. Il existe quatre types de bases azotés, séparés en deux groupes, les pyrimidines (thymine et cytosine), et les purines (adénine et guanine). La liaison entre les deux brins de la double hélice se fait toujours à travers des liaisons A-T et G-C, par des ponts hydrogènes. La thymine et l'adénine sont dites bases complémentaires, de même que la guanine et la cytosine.
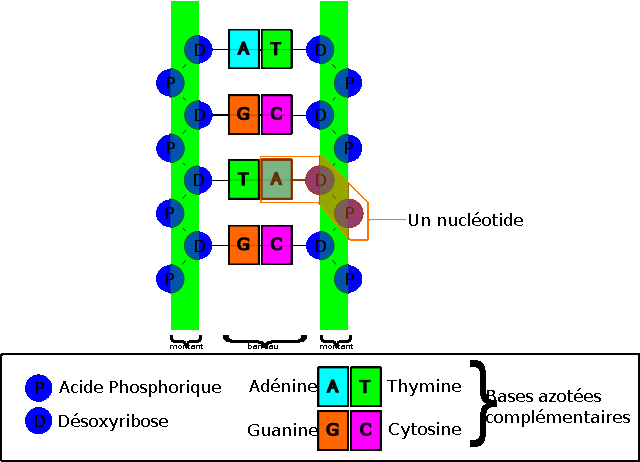
Organisation des nucléotides
La liaison G-C est la plus stable: elle comprend trois ponts hydrogènes.
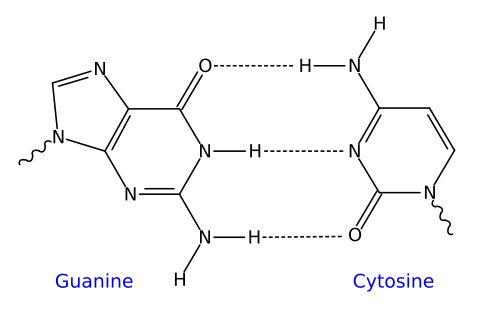
Liaison G-C
La liaison A-T ne comprend elle que deux ponts hydrogène.
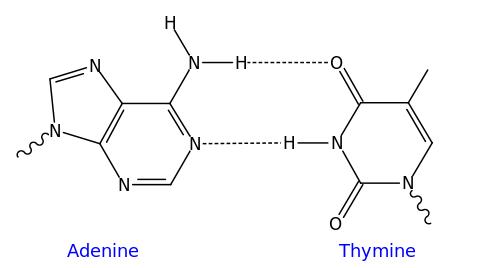
Liaison A-T
L'ADN humain contient environ 3 milliards de paires de base répartis sur les 23 chromosomes. Chaque être humain ayant deux chromosomes de chaque type, notre code génétique complet comprend donc environ 6 milliards de paires de bases. Le nombre de paires de bases par chromosomes est variable, celui en comportant le plus est le chromosome 1 (240 millions de paires de bases, noté 240 Mbps), celui en comportant le moins est le gène 21 (47Mbps).
La duplication de l'ADN s'opère par séparation des paires de bases complémentaires. et par fixation sur chaque demi-brin d'un nouveau demi-brin complémentaire.
Duplication de l'ADN
On obtient ainsi deux brins identiques chacun étant composé d'un demi-brin du segment d'ADN original et d'un nouveau demi-brin synthétisé. On parle de processus semi-conservateur.
Drew Berry, DNA animation, The Walter and Eliza Hall Institute
of Medical Research.
(C) 2007 Howard Hughes Medical Institute
Sur l'animation ci-dessus, on voir comment la double-hélice d'ADN est dépliée dans l'hélicase puis répliquée.
Le fonctionnement précis de la réplication est relativement
complexe, car l'ADN polymérase qui effectue l'élongation et la
duplication ne peut fonctionner que dans un sens. Un des deux demi-brins
(dit direct) est copié en
continu. L'autre demi-brin (dit indirect), est en fait copié
par fragments
(les fragments d'Okazaki qui mesurent environ 200 bases
chez les eucaryotes) comme on le voit dans
l'animation.
La précision de la réplication est de l'ordre de 1 erreur pour 100000
bases. Bien que remarquable, elle est évidemment beaucoup trop faible
si l'on considère le nombre de paires de bases du génome
humain. Cela entraînerait jusqu'à près de 50000 à 100000 erreurs par
division cellulaire. Il existe de nombreux mécanismes de corrections
supplémentaires qui permettent de ramener le taux d'erreur à 1 pour
10.000.000. Néanmoins le taux de mutation n'est jamais nul. Les
mutations sont dangereuses (elles sont par exemple la source de
cancers et de maladies génétiques), mais elles sont aussi
nécessaires à l'évolution pour renouveler le matériel génétique.
Intercalage d'une molécule de benzopyrène
entre deux bases de l'ADN
Le taux d'erreurs, et donc de mutations, devient beaucoup plus important en présence de certains agents qui peuvent être des radiations (comme les UV qui provoquent des liaisons parasites entre deux pyrimidines voisines), ou des produits chimiques comme le benzopyrène (très présent dans le tabac), qui s'intercale entre les bases de l'ADN.
Après avoir vu comment l'ADN se duplique, permettant ainsi la duplication des cellules lors de la mitose et la reproduction lors de la méiose, il nous reste à comprendre comment une cellule est susceptible d'exprimer le code génétique pour fabriquer, entre autres choses, les protéines dont notre corps à besoin. L'opération qui va de l'ADN à la fabrication des protéines comprend plusieurs étapes que nous allons rapidement examiner.
Le code génétique contient un très grand nombre de paires de bases, mais seules certaines séquences spécifiques de ces paires de bases seront décodées et transformées en protéines: ce sont les gènes. Le reste de l'ADN était autre fois appelé junk DNA, ou ADN poubelle. On sait aujourd'hui que même les séquences non-codantes ont une importance dans le fonctionnement de la machinerie cellulaire. On a compris certaines des fonctions de cet ADN (qui joue en particulier un rôle dans l'initiation de la transcription des gènes), d'autres fonctions sont encore peu claires ou mal comprises.
La première étape de décodage de l'ADN est la transcription. Il s'agit de transcrire une séquence de l'ADN (un gène) en une séquence complémentaire de paires de bases azotées, un peu comme lors de la réplication de l'ADN. Mais ici, on ne crée une copie que pour un des deux demi-brins, et cette copie n'est pas de l'ADN, mais de l'ARN (acide ribonucléique).
L'ADN et l'ARN ont des structures chimiques très proches. Les desoxyriboses sont remplacés par des riboses, qui sont globalement les mêmes molécules, avec un atome d'oxygène en plus.
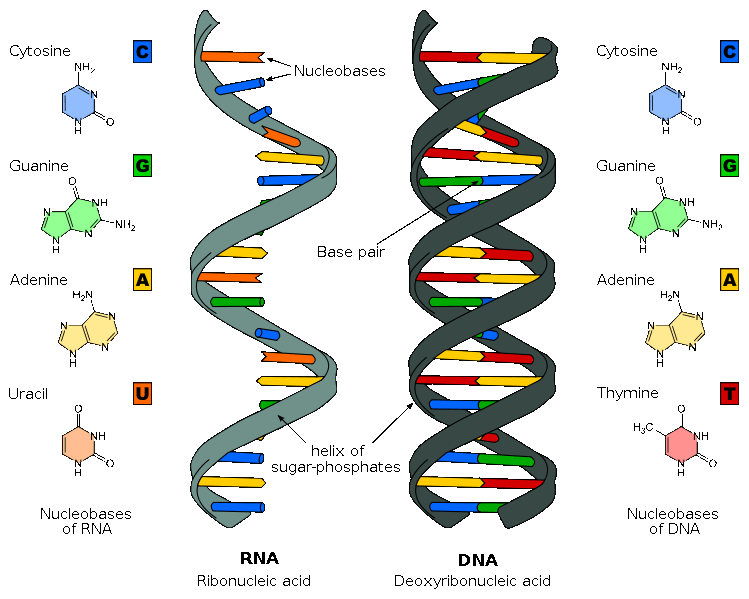
Comparaison de la structure de l'ARN et de l'ADN
Les 4 bases azotées sont presque identiques, la thymine étant simplement remplacée par l'uracile.
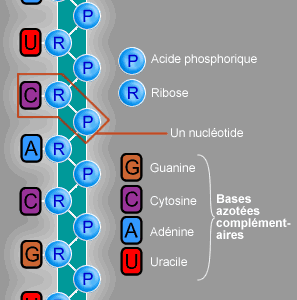
Structure de l'ARN
La transcription consiste donc à séparer temporairement les deux brins de la molécule d'ADN pour créer une copie complémentaire du brin matrice sous la forme d'un brin d'ARN.
![]()
Transcription de l'ADN en ARN
Le déclenchement de l'opération de transcription ne peut se faire que
dans des conditions très spécifiques. Chaque gène est précédé dans le
brin d'ADN d'un ou plusieurs promoteurs, qui permettent à la
machinerie moléculaire qui va assurer la copie de séparer les deux
brins d'ADN et d'initier le processus. Chez les eucaryotes, le
principal promoteur s'appelle la boite TATA. Il s'agit d'une
séquence de 6 bases TATAAA situés 20 à 35 bases avant le
début du gène. Un autre promoteur courant est l'initiateur, une
séquence de 7 bases de la forme (C/T)(C/T)A(C/T/A/G)(C/T)(C/T) [le
signe "/" signifie "ou"], le A se situant au début du site de
transcription.
Il existe bien d'autres promoteurs, comme les boites CAAT et GC, et
bien d'autres facteurs régissant la transcription.
Cette première phase aboutit à la création d'un segment d'ARN dit ARN pré-messager.
Chez les eucaryotes, l'ARN pré-messager doit subir un certain nombre de transformations avant d'être traduit, la plus importante de ces opérations étant l'épissage.
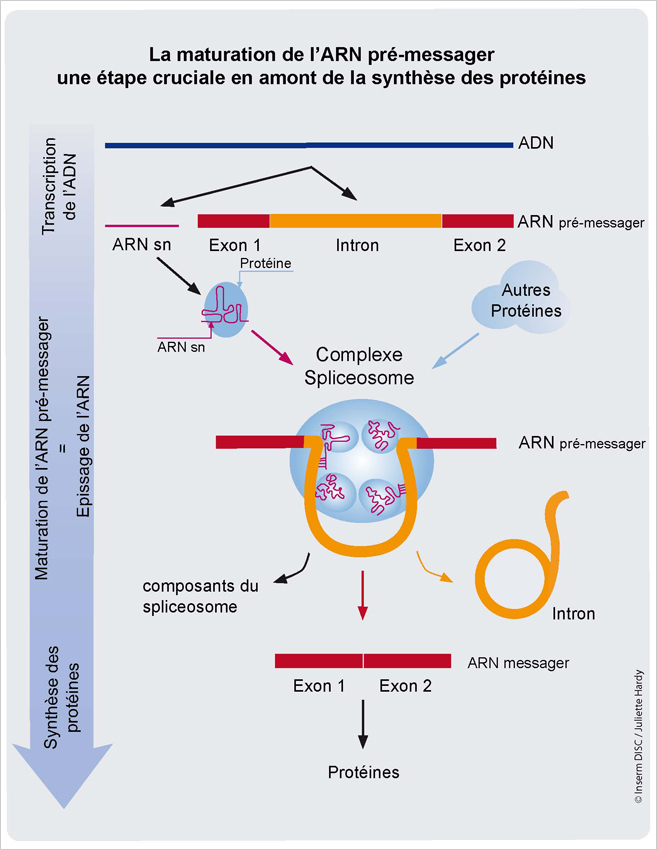
Épissage des introns dans l'ARN pré-messager
Après la transcription, l'ARN pré-messager fabriqué est une suite de paires de
bases. Mais cette suite contient deux types de séquences:
les exons qui seront effectivement traduits, et
les introns, qui doivent être éliminés de la
séquence. L'opération qui élimine les introns s'appelle
l'épissage. Une fois l'épissage
effectué, l'ARN pré-messager est devenu l'ARN messager,
ou ARNm.
L'épissage est une opération complexe qui implique de nombreux
médiateurs: des protéines, mais aussi
un type particulier d'ARN, les ARNsn (small
nuclear ARN). Ces ARNsn sont aussi impliqués dans une
opération de
transcriptase inverse (la transformation d'ARN en ADN) qui
permet sous certaines conditions d'allonger les télomères des
chromosomes, qui raccourcissent normalement à chaque duplication de
l'ADN lors de la mitose. La transcriptase inverse est le mécanisme
utilisé par les rétrovirus pour transformer leur matériel
génétique (qui est codé sous forme d'ARN et non d'ADN) en ADN, ADN qui
est ensuite intégré dans l'ADN des cellules de l'hôte par une enzyme.
L'épissage effectué sur une séquence d'ARN pré-messager peut dépendre de l'environnement cellulaire, on appelle ce phénomène épissage alternatif. Ainsi, un même gène donnera généralement le même ARN pré-messager, mais peut donner naissance à deux ARN messagers différents. Ces ARN messagers différents coderont alors des protéines différentes. Par exemple, le gène codant la protéine CD45 peut être épissé de deux façons donnant pour l'une une forme longue de la protéine et pour l'autre une forme courte de celle-ci, la forme longue étant la seule forme active. La protéine CD45 jouant un rôle dans l'activation des lymphocytes T, la forme courte est produite lorsque ceux-ci ont déjà été activés, afin d'éviter une réponse immunitaire disproportionnée de l'organisme.
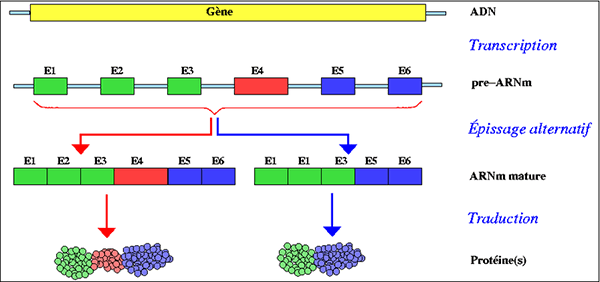
Épissage alternatif
Nous allons nous intéresser à l'étape suivante: la traduction. La traduction est l'opération qui consiste à passer de l'ARNm aux chaînes polypeptidiques (nous parlerons en général de chaînes polypeptidiques plutôt que de protéines, les protéines étant souvent constituées de plusieurs chaînes polypeptidiques qui sont assemblées plus tard, même si la confusion est régulièrement faite).
Les peptides et polypeptides sont des polymères composés d'acides aminés. Les acides alpha-aminés sont composés d'un groupe carboxyle COOH et d'un groupe amine NH, liés entre eux par un atome de carbone qui porte également un atome d'hydrogène et une chaîne latérale R. C'est cette chaîne latérale qui la fonction de l'acide aminé.
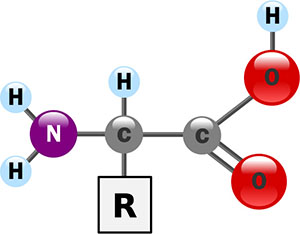
Configuration générale d'un acide alpha-aminé
Les acides aminés polymérisent en libérant une molécule d'eau suivant la réaction chimique:
H2N-CHRa-COOH + H2N-CHRb-COOH -> H2O + H2N-CHRa-CO-NH-CHRb-COOH
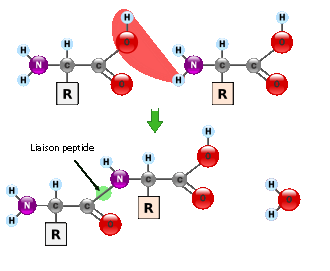
Création d'une liaison peptidique
Ils forment ainsi les ch aines polypeptidiques caractéristiques des protéines (Nous examinerons plus tard les problèmes de configuration spatiale de ces chaînes).
Les acides aminés directement codés par le système génétique sont
environ une vingtaine. Les premiers acides
aminés furent découverts au début du XIXème siècle. On pensa longtemps
qu'il n'existait que 20 acides aminés codés par le système génétique,
avant d'en découvrir un 21ème en 1986, la sélénocystéine, et un 22ème
en 2002, la pyrrolysine, qui ne se rencontre pas chez les
eucaryotes. 19 acides aminés ne contiennent que des atomes
d'hydrogène, d'oxygène et d'azote, 2 (la cystéine et la méthionine)
contiennent un atome de souffre, et la sélénocystéine un atome de
sélénium.
D'autres acides aminés sont utilisés par les organismes vivants, et
construits
par modification post-traductionnelle de la vingtaine d'acides aminés
de base (la citrulline dérive par exemple de l'arginine).
Les 22 acides aminés sont les suivants:
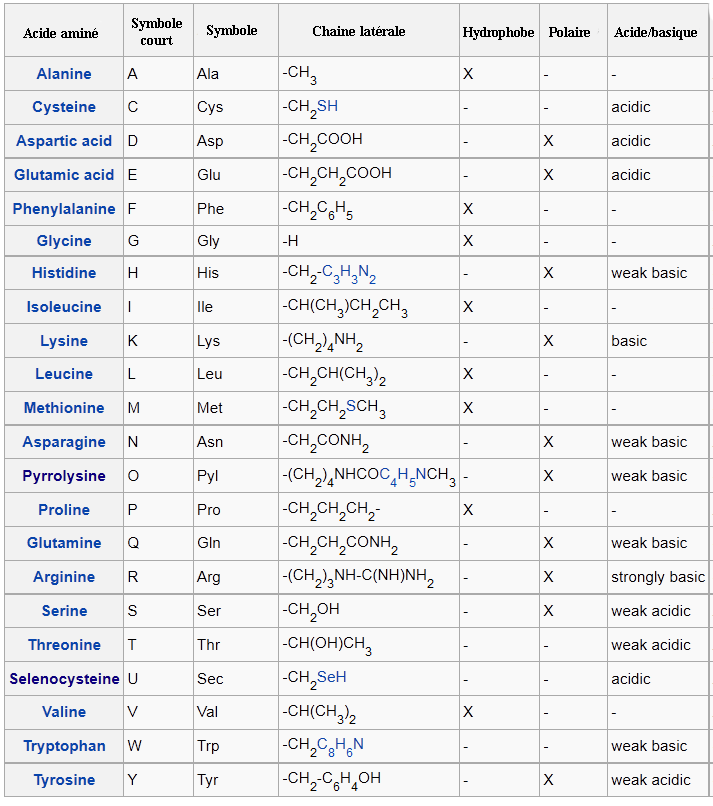
Acides aminés et leurs propriétés
Les acides aminés ont des propriétés variables en fonction de leur chaîne latérale. On distingue 3 caractéristiques principales: hydrophobe/hydrophile, acide/basique et polaire/non-polaire. Ces caractéristiques jouent en particulier sur la configuration spatiale des polypeptides. Les acides aminés hydrophiles, par exemple, ont tendance à se placer à la surface de la protéine, alors que les hydrophobes se retournent vers l'intérieur.
Le but de la traduction est de créer des chaînes polypeptidiques en fonction de la séquence de bases portée par l'ARNm. Le mécanisme général de cette synthèse de protéines est représenté sur la figure ci-dessous.
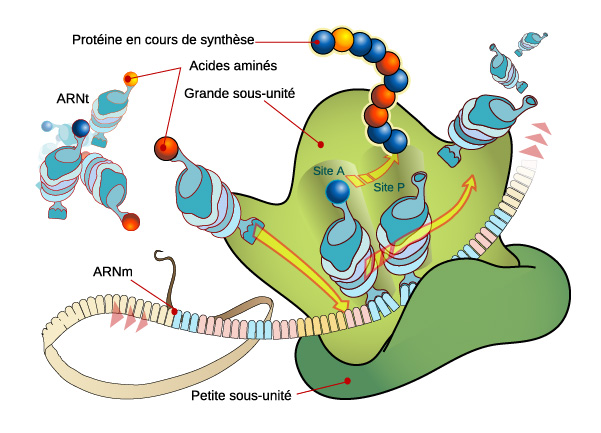
Passage de l'ARNm aux chaînes polypeptidiques
L'opération se déroule grâce aux ribosomes. Les ribosomes sont
constitués de deux sous-unités, appelées sous-unité L (pour large, ou
grande sous-unité), et sous-unité S (pour small, ou petite
sous-unité). Les ribosomes sont composés d'acides aminés et d'ARN,
appelé ARNr, ou ARN ribosomique.
La première étape se déroule à l'intérieur de l'unité S. L'unité S se
charge d'effectuer la liaison entre le brin d'ARN messager et l'ARN de
transfert, ou ARNt.
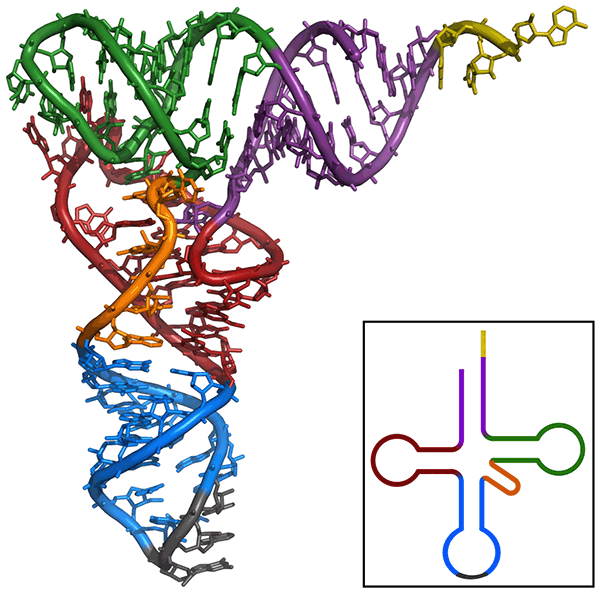
Structure 3D et structure planaire des ARNt
Les ARNt ont une structure dite en "feuille de trèfle", formés de 4 bras. Sur la partie orange se trouve un acide aminé, et sur la partie noire une série de 3 bases azotées appelées anti-codon. Pour une séquence de 3 bases donnée, on trouve toujours le même acide aminé. Le reste de la structure de l'ARNt est identique. Cette séquence de 3 bases est exactement complémentaire d'une séquence de 3 bases se trouvant sur l'ARNm. Pour une séquence de 3 bases données sur l'ARNm, l'ARNt spécifique de cet ARNm va se fixer sur l'ARNm au sein de l'unité S du ribosome.
![]()
Fonctionnement des ARNt
La correspondance entre triplets de bases situés sur l'ARNm et l'acide aminé correspondant s'appelle le code génétique.
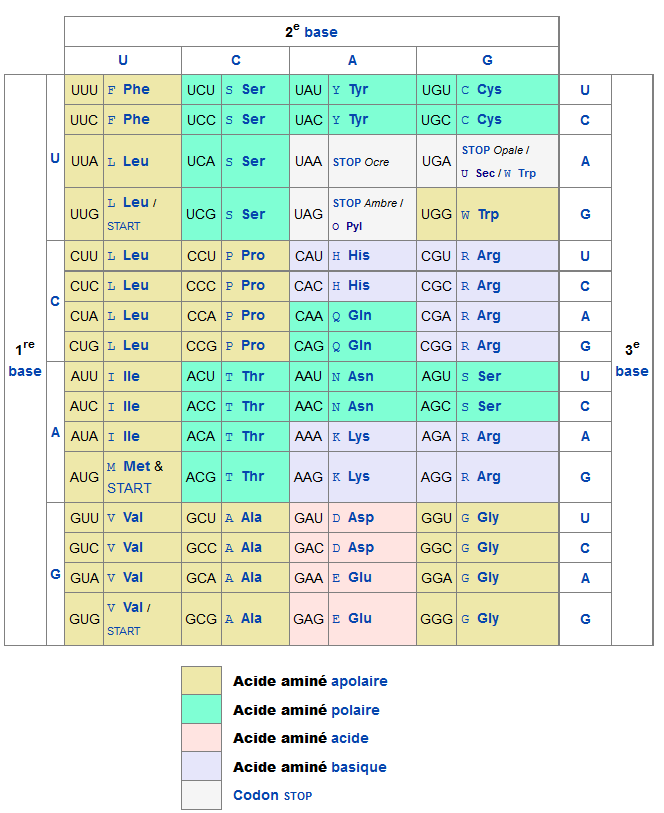
Code génétique
Le code génétique contient exactement 43=64 codons différents. Cependant ce code est dégénéré. Des séquences différentes peuvent en effet coder le même acide aminé. Cette dégénérescence se trouve essentiellement au niveau de la 3ème base du codon. Ainsi, les codons CUU, CUC, CUA et CUG codent tous la leucine. Il existe trois codons particuliers (UUG, AUG et GUG) qui codent respectivement la leucine, la méthionine et la valine mais sont aussi les codons qui se trouvent généralement systématiquement en début de séquence codante (codons START). 3 codons ne codent (en général) aucun acide aminé: ce sont les codons UAA (codon STOP Ocre), le codon UAG (codon STOP Ambre) et le codon UGA (codon STOP Opale). Lorsque le processus de traduction rencontre un de ces codons, la traduction s'arrête. Le code génétique standard représenté ci-dessus ne permet de coder que 20 acides aminés, et non 22. Les deux derniers acides aminés sont codés (sous certaines conditions très particulières) pour la pyrrolysine par le codon STOP UAG et pour la sélénocystéine par le codon STOP UGA.
Après que les ARNt se soient fixés sur l'ARNm, ils traversent l'unité L des ribosomes. Cette unité réalise la polymérisation des acides aminés pour les transformer en une seule chaîne peptidique, et les détache de l'ARNm.
![]()
Passage de l'ARNm aux chaînes polypeptidiques
La chaîne peptidique peut alors quitter le ribosome, puis le noyau de la cellule (voir ci-dessus).
Après synthèse, les chaînes peptidiques connaissent une phase de réarrangement spatial, dite phase de repliement. Cette phase est du aux liaisons hydrogènes qui se créent entre les groupes amines et les groupes carboxyliques des acides aminés.
La forme la plus stable est l'hélice alpha. Il s'agit d'une structure hélicoïdale dont les liaisons hydrogènes assurent la stabilité.
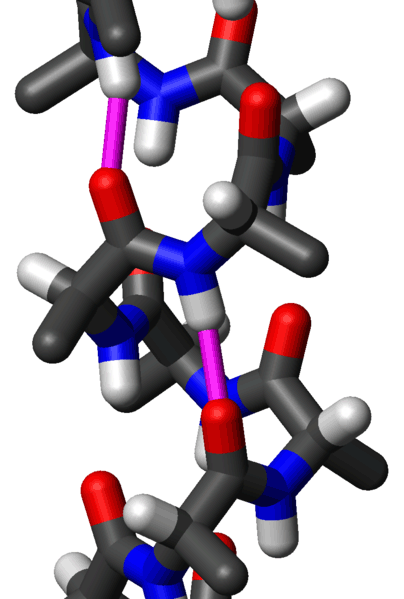
Hélice alpha
Les liaisons hydrogènes entre les groupes NH et CO sont en
violet,
les groupes CO sont en rouge et les groupes NH en bleu.
La seconde structure stable est la structure en feuillet bêta. La chaîne peptidique est repliée sous la forme d'un feuillet relativement rigide.
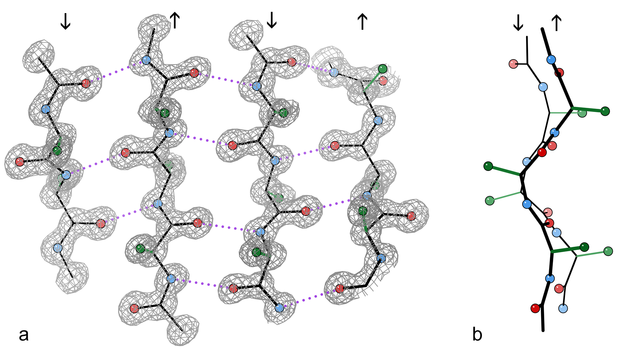
Feuillet bêta
Les liaisons hydrogènes entre les groupements NH et CO sont en
violet,
les atomes d'oxygènes sont en rouge,
les atomes d'azote en bleu.
La plupart des protéines sont des arrangements de plusieurs chaînes
peptidiques. Les protéines dites globulaires, généralement rondes et
solubles dans l'eau (comme l'hémoglobine) sont composées d'un coeur de
feuillets bêta et de plusieurs hélices alpha. Les protéines
structurales, rigides et généralement insolubles dans l'eau (comme la
fibroïne, la protéine sécrétée par le ver à soie), sont
généralement composées essentiellement de feuillets bêta.
La structure spatiale des protéines a une importance considérable sur
leur activité. Ainsi les encéphalites spongiformes transmissibles (la
maladie de Kreutzfeld-Jacob, ou maladie de la vache folle, chez
l'homme) sont dues à des modifications de la structure de certaines
protéines du cerveau. Ces protéines, qui ont normalement une structure
d'hélice alpha, prennent une structure de feuillet bêta lorsqu'elles
sont mises en contact avec des prions. Les prions ne sont
rien d'autres que des protéines ayant elles-mêmes une structure en
feuillet bêta. Il s'ensuit une réaction en chaîne qui finit par
entraîner la transformation de la totalité de ces protéines. Les
maladies à prions ont été le premier exemple de maladies transmissibles
qui ne sont provoquées ni par des bactéries, ni par des virus.
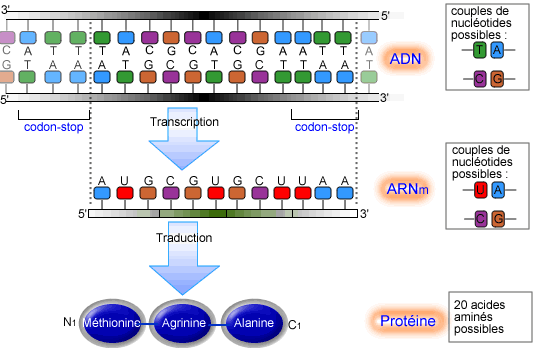
Résumé simplifié des opérations de
transcription/traduction de l'ADN
Le premier séquencement humain génomique a été publié en 2001. Depuis, les méthodes de séquencement intégral ont été raffinées, et l'objectif affiché aujourd'hui est d'être capable de séquencer totalement le génome d'un individu pour un coût de l'ordre de quelques milliers de dollars (l'objectif étant de descendre à 1000$). Ces nouvelles méthodes basées sur du séquencement parallèle massif ont été utilisées pour la première fois en 2008 par les laboratoires de la société Roche pour séquencer le génome de James Watson en 4 mois pour 1.5M$ (à comparer avec les 100M$ nécessaires pour séquencer le génome de Craig Venter en 2007 avec les anciennes technologies).
Les méthodes actuelles de séquencement rapides du génome humain se basent sur le principe suivant: l'ADN est d'abord coupé en fragments de "petite" taille. Ces fragments sont ensuite identifiés suivant diverses techniques, puis réassemblés par des programmes informatiques en tenant compte des zones de recouvrement. Aujourd'hui la taille des fragments varie suivant les technologies, allant de 50 à 200bp pour la technologie de séquencement par synthèse d'Illumina (pour un coût d'environ 10 cents par million de bps séquencés) à 3000bp pour la technologie PacificBio (pour un coût d'environ 2$ par million de paires séquencés).
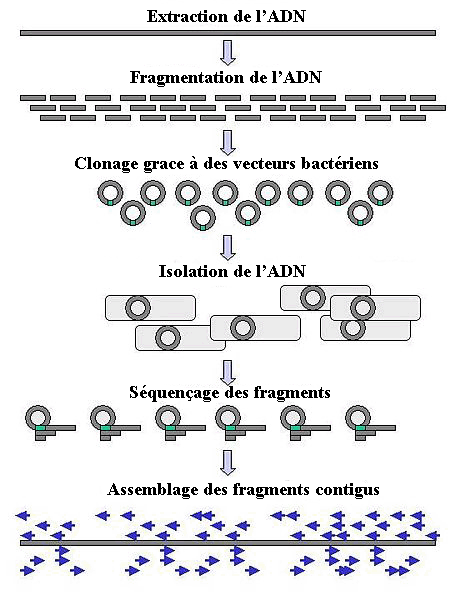
Séquençage de l'ADN
L'assemblage peut se faire suivant deux modes: l'assemblage dit "de novo", qui ne se base sur aucun présupposé, ou l'assemblage par comparaison qui utilise une séquence génétique de référence comme guide. L'assemblage "de novo" est évidemment beaucoup plus coûteux en terme de temps de calcul. L'efficacité et le coût des différentes méthodes dépendent des technologies utilisées, de la taille des fragments de base, etc (les méthodes ayant de très petites tailles de fragments sont par exemple plus complexes à réassembler particulièrement dans les zones du génome présentant des répétitions importantes, etc) De nombreux algorithmes ont été développés pour résoudre ces problèmes.
A la fin de sa vie, Steve Jobs fit séquencer son génome pour 100000$ dans une tentative de combattre un cancer, en faisant comparer l'ADN des cellules normales et celui des cellules cancéreuses, afin de trouver les mutations responsables et possiblement les médicaments les plus susceptibles d'être efficaces contre la maladie. Les techniques de tests génétiques pour identifier les patients à risque et trouver les médicaments le mieux adaptés sont certainement appelées à se développer. En 2010, Stephen Quake, un des inventeurs de la technologie du séquencement rapide, séquença son propre génome. Sa famille étant sujette aux problèmes cardiaques, les résultats furent examinés en détail, et montrèrent une probabilité augmentée de 50% de développer une maladie coronarienne, ainsi qu'une probable bonne réponse aux statines comme traitement. Le séquencement complet du génome est cependant une solution encore chère alors que les méthodes basées sur l'analyse des SNP dont nous parlerons plus loin sont bien moins coûteuses, et suffisantes dans la très grande majorité des cas.
Il n'y a pas aujourd'hui de consensus exact sur le nombre total de gènes dans le génome humain. En effet, si la chaîne complète de nucléotides a été séquencée pour un certains nombres d'individus, on ignore quelles sont exactement les régions codantes et les régions non-codantes. Il existe plusieurs méthodes tentant d'identifier les gènes:
Les méthodes ab-initio ont
tendance à surestimer le nombre
de gènes, les méthodes comparatives à le sous-estimer. On pense que le
nombre total de gènes humains "devrait" être aux alentours de 30000,
ce qui n'est guère que 1/3 de plus que le ver de
terre C. elegans...
Toutes ces méthodes font lourdement
appel à différents domaines de l'informatique comme la
manipulation de grands volumes de données, l'apprentissage,
l'optimisation, etc...
Le "dogme central" de la biologie moléculaire a été énoncé par Francis Crick en 1958. Il peut se résumer par le schéma suivant:
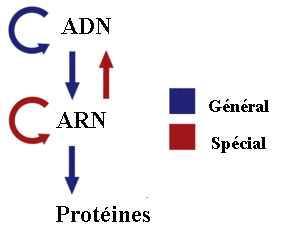
Flux d'information entre ADN, ARN et protéines
La version simplifiée de ce "dogme" (le mot est particulièrement mal
choisi, suivant l'aveu de Francis Crick lui-même) a été énoncée par
Marshall Nirenberg: "L'ADN fait de l'ARN qui fait des protéines" (les deux
flèches bleues descendantes de notre schéma). L'autre flèche bleue
correspond à la réplication de l'ADN. Ce sont les seuls phénomènes
généraux de "transmission" de l'information génétique. La flèche rouge
ARN→ADN correspond à la transcriptase inverse, un modèle utilisé par
exemple par certains virus pour infecter un hôte, la flèche rouge
ARN↔ARN correspond au mode de reproduction des virus qui ne
contiennent que de l'ARN.
Il existe quelques très rares exceptions à ce principe. Nous en avons
vu une avec les prions, qui sont une forme (faible) de transmission de
propriétés de protéine à protéine (la modification
post-translationnelle d'une protéine en est une autre).
Mais il en existe une plus intéressante
avec les intéines. Une intéine est un
fragment de protéines capables de s'exciser de la chaîne d'acides
aminés et de se (re)lier par une liaison peptidique.
Généralement un seul chromosome est porteur de l'allèle codant ces
intéines, mais celles-ci ont un mécanisme particulier (appelé "homing"
en anglais) qui leur permet de modifier l'autre chromosome pour qu'il
les code à leur tour.
Ces intéines contiennent en effet des endonucléases: les
endonucléases sont des protéines enzymatiques capables de
couper le lien phosphodiester d'une chaîne polypeptidique, par exemple
l'ADN, à un endroit spécifique appelé site de restriction et
identifié par une séquence
palindromique de 4 à 6 bases en général. Dans le cas des intéines, il
s'agit d'endonucléases particulière, les méganucléases, capables de
reconnaître des sites de restriction particulièrement longs,
correspondant précisément au gène contenant l'allèle ne codant pas
l'intéine.
La méganucléase une fois libérée va rejoindre
ce site et le couper,
activant ainsi le
mécanisme de réparation de l'ADN qui va réparer le chromosome en
copiant l'allèle de l'autre chromosome, allèle qui portait la mutation
fabriquant l'intéine.
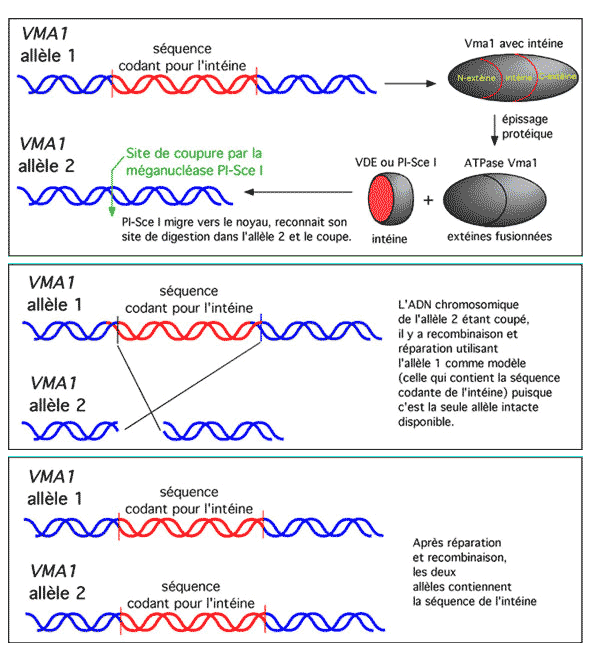
Mécanisme de modification de l'ADN par la méganucléase PI-Scel
Extrait du site de Benoît Leblanc (Université d'Usherbrooke)
Il s'agit d'un exemple de comportement "égoïste" ("selfish") permettant à une protéine de modifier la séquence d'ADN pour assurer sa production.
La notion de dominance a été introduite par Mendel bien avant que l'on connaisse les mécanismes de transmission de l'information génétique. Elle continue a être utilisée largement même si, comme nous allons le voir, il serait plus utile d'y renoncer pour s'intéresser aux conséquences phénotypiques des interactions entre allèles, tant cette notion de dominance est une sur-simplification qui peut se révéler trompeuse ou inadaptée dans de nombreux cas.
Les expériences de Mendel sont bien connus et nous ne les rappellerons que brièvement. Mendel va étudier différentes caractéristiques du pois, rappelées dans le tableau ci-dessous.
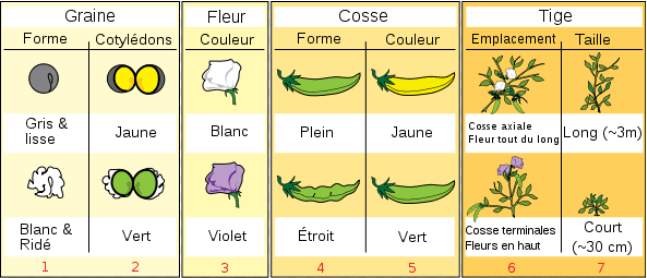
Différentes caractéristiques étudiées par Mendel
Mendel va d'abord étudier une seule caractéristique du pois, par exemple l'aspect, en partant de lignées pures de pois lisses et de pois ridés. Il va donc croiser des plantes de type lisse avec des plantes de type ridé et constater que tous les éléments de la première génération (appelée F1) sont de type lisse.
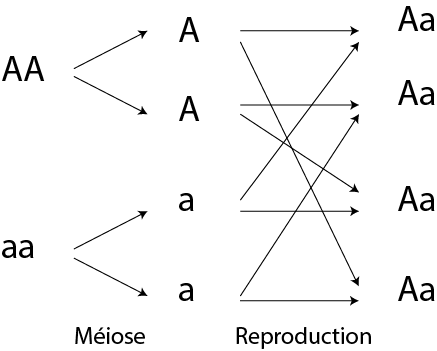
Croisement F1
Le caractère [lisse] est donc dominant par rapport au caractère [ridé]. En croisant les membres de la génération F1 entre eux, il obtient en revanche en F2 25% de pois ridés et 75% de pois lisses.
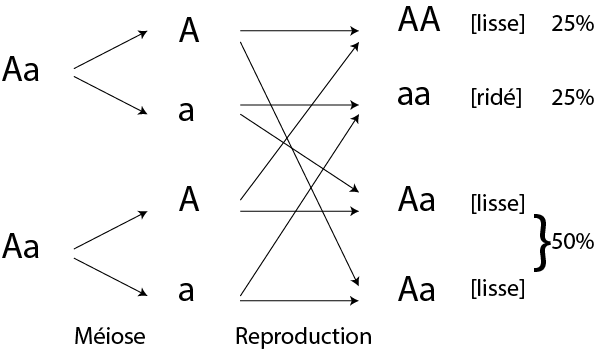
Croisement F2
Cela est bien cohérent avec un chromosome diploïde, dont l'allèle codant le caractère lisse (noté A) est dominant, et dont l'allèle codant le caractère ridé (noté a) est récessif. Les individus [lisse] purs sont de type AA, et les individus [ridé] purs de type aa. En première génération, on obtient exclusivement des génotypes Aa qui n'expriment qu'un phénotype [lisse]. En revanche, en génération F2 on obtient statistiquement 25% de AA, 50% de Aa et 25 % de aa, soit phénotypiquement 25% de [ridé] et 75% de [lisse].
Mendel testera par la suite deux caractères simultanément, comme la forme et la couleur, sachant que le caractère [jaune] est dominant par rapport au caractère [vert]. Il croisera deux lignées pures, l'une de pois [lisse/jaune] avec les allèles dominants AB et une lignée pure [ridé/vert] avec les allèles récessifs ab. En génération F1, on obtient exclusivement des pois [lisse/jaune], avec deux distributions indépendantes (hypothèse d'indépendance des caractères) de (3/4 [lisse],1/4 [ridé]) et (3/4 [jaune],1/4 [ridé]) ce qui donnera en génération F2 une répartition de 9/16 de [lisse/jaune], 3/16 de [lisse/vert], 3/16 de [ridé/jaune] et 1/16 de [ridé/vert], suivant le ratio 9:3:3:1.
Mendel posera les bases de la génétique à partir de ces expériences. Mais il s'agit de cas "idéaux" comme nous allons le voir.
Les expériences de Mendel semblaient montrer une indépendance totale entre les différents caractères. Mais quelques années plus tard William Bateson et Reginal Punnet vont montrer que cette hypothèse est parfois incorrecte. En travaillant sur le pois de senteur, ils étudient deux caractères, la couleur de la fleur qui peut être violette (V, dominant) ou rouge (v, récessif), et la forme des grains de pollen qui peuvent être longs (L, dominant) ou ronds (l, récessif). En croisant des lignées pures VVLL et vvll, ils s'attendaient à obtenir le ratio classique 9:3:3:1. Mais le résultat fut un peu différent.
| Phénotype et génotype | Observé | Attendu |
| Violet,long (VvLl) | 284 | 216 |
| Violet,rond (Vvll) | 21 | 72 |
| Rouge,long (vvLl) | 21 | 72 |
| Rouge,rond (vvll) | 55 | 24 |
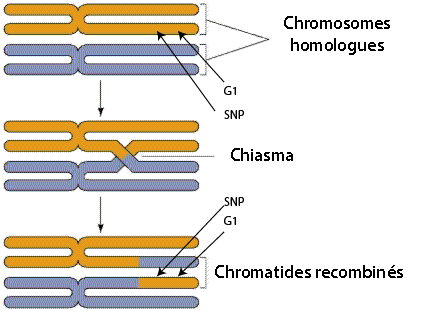
Il est clair qu'ici il existe une liaison entre les deux gènes puisque la présence d'occurrence de P avec L et de p avec l est bien plus importante que celle prévue par le ratio théorique. L'explication est simple: les deux gènes se trouvent sur le même chromosome et sont relativement proches.
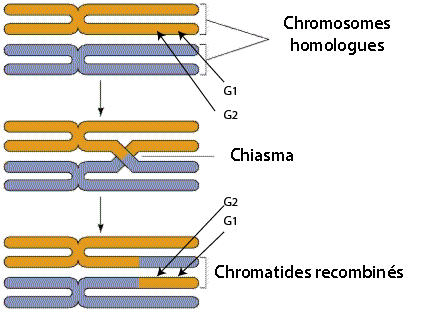
Recombinaison qui maintient la liaison entre les gènes G1 et G2
Lorsque les deux gènes sont proches sur le chromosome, la probabilité que la recombinaison coupe le chromosome entre les deux gènes est beaucoup plus faible que 1/2 et les deux gènes ont de grande chance de rester liés après la recombinaison. En fait, les analyses statistiques de ce type furent utilisées très tôt pour construire des cartes génétiques permettant d'identifier la position relative des gènes sur les chromosomes.
L'épistasie est une interaction entre deux gènes situés à des emplacements différents, qui est parfois confondu avec une dominance. L'épistasie modifie le ratio classique 9:3:3:1 que l'on observe avec deux gènes non épistatiques. L'épistasie est observée dans la grande majorité des organismes vivants et implique dans le cas général beaucoup plus de deux gènes. Pour deux gènes il existe 14 interactions épistatiques possibles.
Un exemple d'épistasie récessive se présente par exemple quand l'un des gènes détermine si le pigment de la fleur sera jaune (AA ou Aa) ou vert (aa), alors que l'autre gène détermine si un pigment sera produit (BB ou Bb) ou si aucun ne sera produit (bb), donnant alors une fleur blanche. On dit alors que le gène B présente une épistasie récessive par rapport au gène A: lorsque B est homozygote pour l'allèle bb, il supprime toute expression du gène A. Lors du croisement de deux plantes AaBb (qui sont jaunes) on observera un ratio 9:3:4 de fleurs jaunes:vertes:blanches.
Dans le cas de l'épistasie dominante, la couleur de la plante est toujours codé par le gène A comme ci-dessus, mais pour que le pigment soit fabriqué il faut qu'un précurseur spécifique soit produit, la production de ce précurseur dépendant d'un gène D. Le précurseur est produit si et seulement si le gène est un homozygote dd. Lors du croisement de plantes AaDd (qui sont blanches), on observera un ratio 12:3:1 de plantes blanches:jaunes:vertes.
Il ne s'agit là que de deux exemples d'épistasie avec deux loci. Il en existe beaucoup d'autres, sans évidemment mentionner les cas impliquant (beaucoup) plus de deux gènes lors de l'expression de phénotype, cas qui sont pourtant plus la règle que l'exception en général.
Pour bien comprendre le phénomène de codominance, il faut bien se rappeler le "dogme fondamental". L'ADN produit de l'ARN qui produit des protéines. Dans le cas de la codominance, nous nous trouvons face à un cas où les deux allèles du gène sont susceptibles de coder une protéine valide, mais pourtant différente. C'est par exemple le cas du gène codant les antigènes sanguins. Il existe trois allèles possibles: Ia qui code l'antigène A, Ib qui code l'antigène B, et Io qui ne produit aucun antigène (il est récessif par rapport aux deux autres). On se retrouve donc face à plusieurs cas de figure:
| Allèles | IaIa | IaIb | IaIo | IbIb | IbIo | IoIo |
| Groupe sanguin | A | AB | A | B | B | O |
Dans ce cas, deux parents porteurs l'un des allèles IaIo (groupe A) et l'autre des allèles IbIo (groupe B) auront statistiquement 25% de chance d'avoir un enfant de groupe A, 25% de chance d'avoir un enfant de groupe B, 25% de chance d'avoir un enfant de groupe AB et 25% de chance d'avoir un enfant de groupe O.
La codominance peut aussi s'observer dans bien d'autres cas, comme celui du rhododendron, pour lequel le croisement d'une plante blanche et d'une plante rouge donne une plante présentant des zones blanches et rouges.

Rhododendron exprimant une codominance blanc/rouge
Il commence à être à peu près clair que la notion de dominance est plus flou que ne le croyait Mendel, et qu'elle est étroitement lié à la capacité d'un allèle à produire (ou ne pas produire) certaines protéines. En fait les cas simples de dominance se produisent lorsque l'un des allèle est non fonctionnel (il n'est pas transcrit, ou sa séquence génétique est incorrecte), et lorsque le fonctionnement d'un seul allèle suffit à produire suffisamment de protéines pour que le phénotype d'un individu hétérozygote soit identique à celui d'un individu homozygote pour l'allèle fonctionnel. C'est par exemple le cas du phénotype albinos chez l'être humain (même si la réalité est là aussi un peu plus complexe). Mais il existe de nombreux cas intermédiaires. Nous allons résumer cela:
Nous allons maintenant examiner en détail le cas d'une maladie génétique, l'anémie falciforme, qui se trouve précisément dans la "zone grise" en matière de dominance.
L'hémoglobine est une protéine complexe composée de quatre chaînes polypeptidiques et de quatre sites (appelé hèmes) recevant un ion ferreux sur lequel peut se fixer un ion oxygène. Sa fonction est de transporter l'oxygène, mais aussi d'autres gaz comme le gaz carbonique.
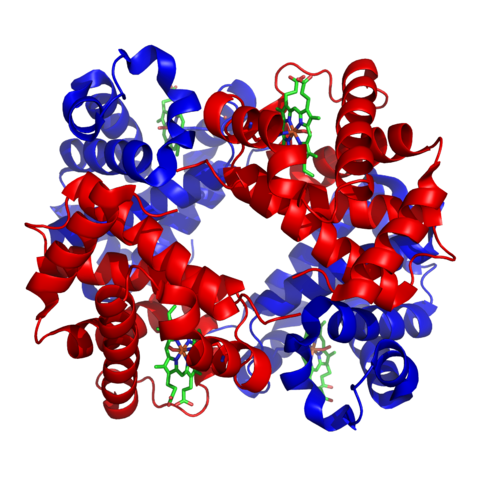
Structure de l'hémoglobine A
Les chaînes alpha sont en rouge, les chaînes bêta en bleu
L'hème recevant la molécule de fer est en vert
Il existe plusieurs types d'hémoglobine, dont la concentration varie dans le corps humain en fonction du stade de développement. A la naissance, l'hémoglobine F (composée de deux chaînes peptidiques alpha et de deux chaînes peptidiques gamma) est la plus importante, mais dès l'âge de 6 mois, environ 95% de l'hémoglobine est de l'hémoglobine A, composée de deux chaînes peptidiques alpha et de deux chaînes peptidiques bêta (il existe d'autres hémoglobines comprenant d'autres chaînes, au stade embryonnaire et au stade foetal).
Les cinq gènes codant les chaînes bêta, delta, gamma A, gamma G et epsilon se trouvent toutes positionnés dans la même zone (appelé zone HBB@) du chromosome 11. (Les chaînes alpha sont synthétisées par un gène situé sur le chromosome 16.)

Séquence des nucléotides codant les cinq gènes
Ordre des gènes: HBB, HBD, HBG1, HBG2 et HBE1
Nous allons nous intéresser ici à une mutation du gène HBB codant la chaîne bêta de l'hémoglobine. Cette mutation se situe à la vingtième position de la zone bêta, et ne concerne qu'un seul nucléotide (le vingtième), et transforme une adénine (A) en thymine (T). Elle change ainsi un codon GAG en codon GTG.
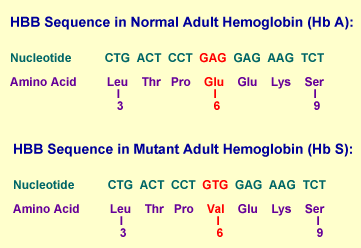
L'acide glutamique (polaire) codé par GAG est remplacé par une valine (non polaire et hydrophobe) codée par GTG. L'hémoglobine formé avec cette chaise bêta modifiée est appelée hémoglobine S. La modification des propriétés chimiques va entraîner tout un ensemble de modifications phénotypiques, dont la plus significative est la possibilité pour la protéine de polymériser lorsqu'elle ne porte plus d'atome d'oxygène. Le globule rouge se déforme alors pour prendre la forme caractéristique d'une faucille, d'où le nom de la maladie, anémie falciforme, parfois appelé aussi drépanocytose.
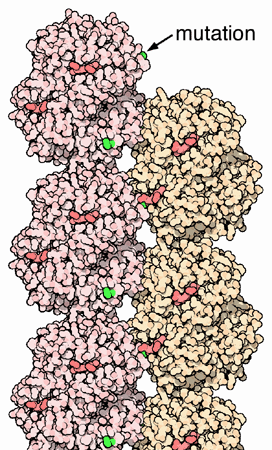
Polymérisation de la forme désoxygénée de l'hémoglobine S
Les protéines d'hémoglobine S ont une durée de vie beaucoup plus courtes (une dizaine de jours contre plus du triple pour l'hémoglobine A), entraînant ainsi une anémie. D'autre part, la polymérisation des globules rouges peut aussi entraîner l'occlusion de petits vaisseaux sanguins et provoquer ainsi des douleurs et des lésions plus ou moins importantes au niveau des bras, des jambes et de certains organes (rate, reins, cerveau), avec des effets secondaires qui peuvent être létaux.
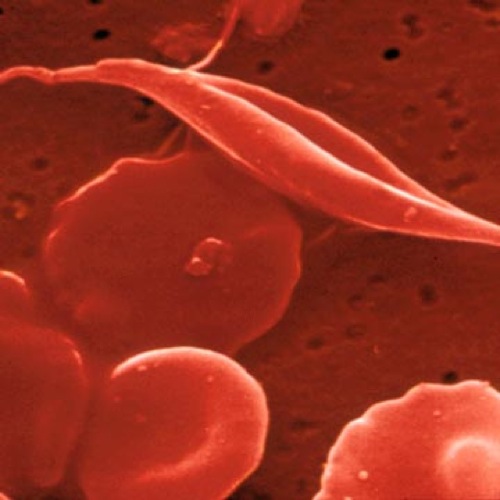
Globules rouges normal et falciforme
La maladie est potentiellement létale pour les seuls porteurs homozygotes de l'allèle muté. Les porteurs hétérozygotes sont en apparence sains, mais ils peuvent cependant déclencher certains symptômes de la maladie dans des circonstances particulières: efforts violents, déshydratation ou manque d'oxygène (lié à l'altitude par exemple). En France, Lassana Diarra fut privé de la coupe du monde en Afrique du Sud (en altitude) pour cette raison. Aux Etats-Unis le joueur de football américain Ryan Clark a été victime d'un infarctus de la rate pendant un match à Denver au stade d'Invesco field at Mile High (qui, comme son nom l'indique, se trouve à environ 1600m d'altitude).
La majeure partie des porteurs de la mutation sont originaires d'Afrique, ou de régions limitrophes. En effet, l'hémoglobine S est beaucoup moins sensible au paludisme que l'hémoglobine A et les porteurs, même hétérozygotes, de la mutation sont partiellement protégés de la maladie. On pense qu'environ un afro-américain sur douze serait porteur d'un gène HBS.
La drépanocytose est l'exemple d'une maladie pour lequel aucun des deux allèles n'est véritablement dominant. Les porteurs hétérozygotes ne sont ni totalement identiques (au sens phénotypique) aux porteurs homozygotes HBA, ni identiques aux porteurs homozygotes HBS.
Une fois de plus il est donc bon de rappeler que la notion de
dominance doit bien souvent être nuancée, et qu'il s'agit bien d'une
notion floue et non d'une notion binaire.
Le cas est encore plus
frappant pour certaines maladies comme la phénylcétonurie (PKU) qui
est un
trouble de la métabolisation de la phénylalanine, un acide aminé
d'origine alimentaire qui doit être métabolisé par une enzyme du foie,
la phénylalanie
hydroxylase (PAH). Le gène de la PAH est situé sur le chromosome 12,
et il présente de nombreuses formes alléliques (A, B, C,...). Un
porteur sain (AA) aura une fabrication de 100% de PAH, un porteur AB
de 30%, un porteur BB de 0.3%, un porteur CC de 5% etc, entraînant des
formes plus ou moins marquées de la maladie. Si la maladie est non
traitée, l'excès de phénylalanine peut entraîner de graves retards de
développement mental.
Nous allons nous intéresser dans le paragraphe suivant à des mutations portées par les chromosomes non-autosomiques. En ce qui concerne les chromosomes autosomiques il est bon de retenir que les maladies génétiques graves purement dominantes sont très rares, car étant généralement fatales au porteur d'un seul gène défectueux, elles ne peuvent se transmettre à la génération suivante, contrairement aux maladies récessives, qui peuvent être transmises par un porteur hétérozygote "sain" au sens du phénotype. La maladie de Huntington sur-citée est un exemple un peu exceptionnel, car elle ne se déclenche en général que tardivement (vers une quarantaine d'années).
Un exemple de mutation autosomique "dominante" SNP d'une seule base entraînant une maladie génétique grave est la mutation du gène ACVR1 (chromosome 2, bras long) où une guanine en position 206 est remplacée par une adénine, transformant un codon CGC en codon CAC, et remplaçant ainsi dans la protéine codée une arginine par une histidine. La maladie associée appelée fibrodysplasie ossifiante progressive, ou plus communément maladie de l'homme de pierre, entraîne la transformation de tous les muscles squelettiques (à l'exception de ceux du coeur et du diaphragme), des tendons et des ligaments en plaques osseuses. Cette maladie orpheline extrêmement rare est généralement provoquée par une mutation "de novo".
Nous allons revenir brièvement dans ce paragraphe sur le cas particulier de la transmission des caractères génétiques par les chromosomes sexuels. Nous ne nous intéresserons ici qu'au système de détermination du sexe dit XY (globalement celui des mammifères, quelques insectes comme la drosophile, et quelques plantes, comme le Gingko).
Il existe principalement deux autres systèmes de détermination de sexe:
Il existe bien des variantes de ces systèmes qu'il serait trop long (et sans grand intérêt) de détailler ici.
Les chromosomes X et Y n'ont évidemment pas un comportement symétrique, puisque seuls les mâles ont un chromosome Y (avec un chromosome X), alors que les femelles disposent de deux chromosomes X.
Le chromosome Y comporte environ 50 millions de paires de base. L'ADN est passé directement de père en fils, permettant d'établir facilement des données de filiation paternelle, et ce sur de très longue durée (comme l'ADN mitochondrial pour les femelles). Le chromosome Y ne se recombinant pas, la dérive génétique a entraîné au fil de l'évolution (la différentiation XY aurait eu lieu il y a environ 170 millions d'années) la perte d'environ 95% des gènes contenus initialement dans le chromosome Y. Il semble cependant qu'il n'y aurait eu aucune perte de gène depuis la divergence humains-chimpanzés il y a environ 6 à 7 millions d'années, ce qui tendrait à prouver que la perte des gènes n'est pas linéaire.
Le chromosome Y porte peu de gènes (environ 80), et ces gènes ne sont pas
fondamentaux pour la vie, puisque la moitié de l'humanité fonctionne
parfaitement sans eux. La majorité des désordres génétiques graves
sont liés aux caractères masculins directement codés par Y. Ainsi, les gènes
de la région AZF (AZF1, AZF2, USP9Y, etc) lorsqu'ils sont dis-fonctionnels,
entraînent généralement une infertilité masculine, et de ce fait ne se
transmettent pas. Un cas plus complexe est celui du gène SRY (Sex
determining Region Y); ce gène code la protéine SRY qui est
l'initiateur de la différentiation mâle/femelle. Un gène SRY non
fonctionnel pour un individu XY entraînera un phénotype
majoritairement femelle, au moins jusqu'à la puberté (syndrome de
Swyer); par la suite, l'absence de gonades fonctionnelles empêchent
la fabrication d'hormones sexuelles, et les caractères sexuels
secondaires n'apparaissent pas à la puberté.
Réciproquement, lorsque
le gène SRY se
déplace sur un chromosome X lors de la méiose (translocation), on a
des individus de caryotype XX présentant des caractéristiques
essentiellement mâles (syndrome de La Chapelle).
Le gène SRY avait été proposé comme moyen de distinguer les
hommes des femmes dans les compétitions sportives (en particulier aux
jeux olympiques). A la suite de nombreuses polémiques, cette méthode a
été abandonnée.
La transmission des caractères et des gènes par le chromosome Y
porte le nom d'hérédité holandrique .
Le chromosome X contient environ 150 millions de paires de bases et 2000 gènes. Le chromosome X contient peu de gènes déterminant les caractères sexuels, la majorité de la détermination sexuelle étant codée par le chromosome Y (ou par son absence). Les mâles possédant un seul chromosome X et les femelles deux chromosomes X, l'évolution a mis en place un mécanisme d'inactivation pour éviter la production en quantité double des protéines codés par les gènes du chromosome X chez les femelles. Ce phénomène est parfois appelé Lyonisation, du nom de son découvreur Mary Lyon.
En dehors des marsupiaux, où le chromosome inactivé est toujours le chromosome X paternel, l'inactivation d'un chromosome X est aléatoire chez les mammifères. En revanche, une fois celui-ci inactivé dans une cellule, tous les descendants de cette cellule garderont le même chromosome inactivé (noté Xi en général, par opposition à Xa pour le X actif). Cette inactivation d'un chromosome X aboutit à des manifestations qui ressemblent à du mosaïcisme, et qui ne se présentent que chez les femelles.
Un des exemples les plus visuels est la coloration des chattes "écaille de tortue". Chez ces chats, la couleur orange est liée à un gène situé sur le chromosome X. Les mâles n'ayant qu'un chromosome X, les mâles ont soit le phénotype orange, soit ne l'ont pas, et il n'y a pas de chats mâles tricolores, sauf ceux (1 sur 3000 environ) souffrant du syndrome de Klinefelter, c'est à dire des chats ayant un caryotype XXY, et présentant des caractéristiques essentiellement mâles du fait de la présence d'un chromosome Y. Chez les femelles hétérozygotes, la coloration orange va apparaître par tâches correspondant aux groupes de cellules activant l'un ou l'autre des chromosomes X.

Conséquences de l'inactivation aléatoire d'un
chromosome X sur une chatte "écaille de tortue"
Comme les individus mâles n'ont qu'un seul chromosome X, la transmission des maladies liées au chromosome X est très différentes de celles liées aux chromosomes autosomes (il est par exemple impossible pour un individu mâle de transmettre une maladie génétique liée au chromosome X à des descendants également mâles). Nous allons voir quelques exemples très caractéristiques ci-dessous.
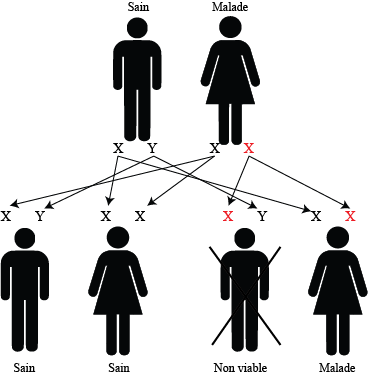
Exemple de transmission de maladie dominante liée à l'X
Les enfants mâles viables sont sains, une fille sur deux est malade
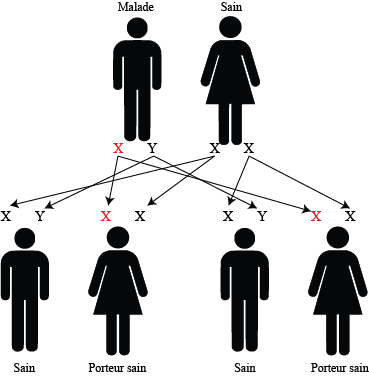
Un père atteint, une mère homozygote saine
100% des garçons sont sains,
100% des filles sont porteurs sains
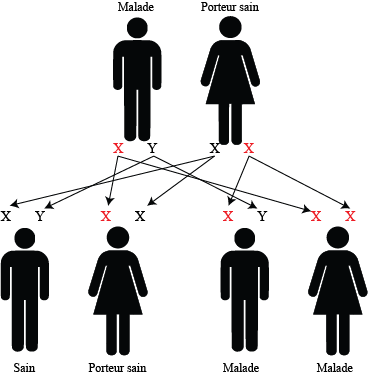
Un père atteint, une mère porteur sain
50% des garçons sont atteints, 50% sont sains
100% des filles sont porteurs sains
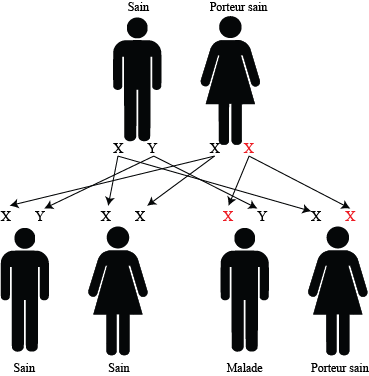
Un père sain, une mère porteur sain
50% des garçons sont atteints, 50% sont sains
50% des filles sont saines, 50% porteurs sains
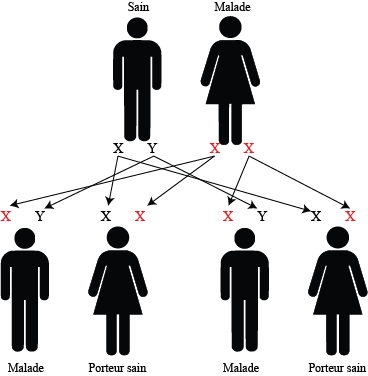
Un père sain, une mère atteinte
100% des garçons sont atteints
100% des filles sont porteurs sains
On remarquera que, pour une anomalie génétique récessive non létale liée à l'X, si la probabilité de trouver dans une population un individu mâle atteint est de p, la probabilité de trouver un individu femelle atteint ne sera que de p2. Pour les anomalies récessives plus graves, les individus mâles ne se reproduisant généralement pas, il n'existe jamais d'individu de sexe féminin atteint. Les maladies récessives liées à l'X se rencontrent donc essentiellement chez les hommes.
L'invalidation du chromosome X est un phénomène un peu plus complexe qu'indiqué ci-dessus. D'une part, tous les gènes ne sont pas inactivés sur le chromosome Xi: le gène Xist (de même que les gènes Jpx, Ftx, Tsx et Cnbp2) n'est activé que sur le chromosome Xi. Il s'agit d'un gène d'ARN non codant: il code un ARN qui n'est pas traduit en une protéine (comme les gènes codant les ARN de transfert ou les ARN ribosomaux). Cet ARN transcrit entoure le chromosome Xi, ce qui est essentiel à son inactivation. D'autre part, certaines régions du chromosome X dites pseudo-autosomales sont similaires à des régions du chromosome Y, et ces régions du chromosome Xi restent actives (ce qui permet d'avoir un dosage équilibré entre individus XY et XX). On sait que chez les personnes possédant un X additionnel (caryotype XXX pour le syndrome triple-X), les deux X additionnels sont inactivés, mais toutes les zones X pseudo-autosomales restent actives, et ce sont les causes des modifications phénotypiques associées. Dans le cas du syndrome de Turner (caryotype X0), c'est l'absence de l'activité des zones pseudo-autosomales du second chromosome X qui semble être une des causes des manifestations phénotypiques. Les anomalies phénotypiques sont variables: les caryotypes XXX ne sont parfois jamais diagnostiqués, tant leur manifestations peuvent être parfois modérées. Le syndrome de Turner entraîne généralement la stérilité chez les femmes qui en sont atteintes.
Les problèmes de vision de la couleur ("daltonisme") sont également souvent liés à des mutations situés sur le chromosome X. La vision de la couleur est essentiellement liée à la présence dans l'oeil de cellules appelées cônes contenant des pigments sensibles à certaines longueurs d'onde. L'homme possède essentiellement trois gènes codant trois types de pigments sensibles à trois types de longueur d'ondes: OPN1SW qui code le pigment sensible essentiellement aux longueurs d'onde proche du bleu (OPNSW1: opsin1 short wave sensitive), OPN1MW (Medium Wave) celui sensible au vert et OPN1LW (Long Wave) le pigment sensible au rouge. L'homme a donc une vision dite trichromique.
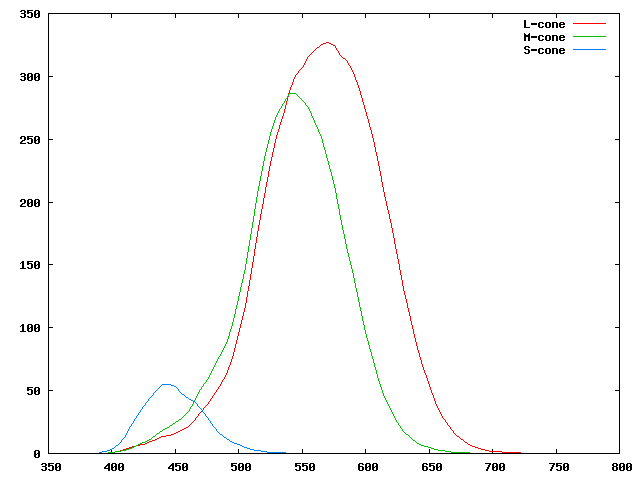
Sensibilité spectrale moyenne des trois types de cône
Extrait de
photo-lovers.org.
Le gène OPN1SW est porté par un chromosome autosome (chromosome 7), et
les problèmes de vision dans le bleu sont donc transmis de façon
autosomique. En revanche, les gènes OPN1MW et OPN1LW, responsables de
la vision rouge/vert, se trouvent sur
le chromosome X et suivent un schéma de transmission récessive liée à
l'X, comme vu ci-dessus. La protanomalie (déficience du gène OPN1LW,
mauvaise vision dans le rouge) se
rencontre chez 1% des hommes et 0.01% des femmes. La deutéranomalie
(déficience du gène OPN1MW, mauvaise vision dans le vert) se rencontre
chez 6% des hommes et 0.4% des femmes.
D'autres gènes (CNGA3, CNGB3, GNAT2) portés par des chromosomes
autosomes interviennent également dans les problèmes de vision de la
couleur.
Il existe un certain nombre de tests permettant de détecter les
anomalies de la perception de la couleur, dont les classiques planches
d'Ishihara, dont on peut voir un exemple ci-dessous.

Test de la discrimination vert/rouge
Un individu non-affecté lira "73"
Les gènes OPN1W et OPN1LW connaissent également des mutations
moins graves modifiant seulement la perception de la couleur associée
au pigment
codé. Cela ne change rien pour les individus mâles, qui conservent une
vision trichromique. En revanche, pour les individus de sexe féminin
qui sont hétérozygotes pour les gènes en question, on peut trouver
plus de trois types de récepteurs, ce qui permettrait une vision
tétrachromique, permettant une meilleure distinction des couleurs.
Pour les gens intéressés par les problèmes de vision de la
couleur, ma page sur la perception de la couleur et la colorimétrie
est disponible
ici.
La régulation de l'expression des gènes est fondamentale dans l'évolution du vivant. C'est un phénomène extrêmement complexe que nous ne ferons qu'aborder très rapidement à titre d'exemple uniquement chez les procaryotes. Chez les eucaryotes, les processus mis en jeu sont encore plus complexes.
On sait depuis les années 1900 que lorsque du glucose est présent dans l'environnement de E. coli, celle-ci synthétise des enzymes nécessaires à la métabolisation du lactose. Inversement, lorsque le lactose est absent, ces enzymes ne sont pas synthétisés.
Les enzymes synthétisées seulement en présence d'un substrat spécifique sont appelés enzymes adaptatives ou inductibles (on parlera aussi de processus inductible). Par opposition, les enzymes synthétisés systématiquement sont appelés enzymes constitutives. Il existe également certaines enzymes qui cessent d'être produites en présence d'un substrat. C'est le cas par exemple du tryptophane, qui peut être synthétisé par les cellules bactériennes, mais cessera de l'être s'il en existe suffisamment dans l'environnement. On dit qu'il s'agit alors d'un processus répressible.
Le contrôle de la régulation, que celle-ci soit inductible ou répressible, peut être positif ou négatif. Le contrôle est négatif quand l'expression du gène se fait sauf si elle est inhibée par une molécule régulatrice. Le contrôle est positif seulement si une molécule régulatrice est présente pour stimuler la production d'ADN.
Nous allons détailler le fonctionnement de la régulation des enzymes de métabolisation du lactose chez E. coli, un travail qui valut à Jacques Monod, François Jacob et André Lwoff le prix Nobel en 1965.
En présence de lactose, E. coli produit des enzymes métabolisant le lactose. Il s'agit donc d'un processus inductible, le lactose étant l'inducteur. Nous allons maintenant voir qu'il s'agit aussi d'un contrôle négatif, puisque l'expression du gène se fait sauf si elle est inhibée par une molécule régulatrice.
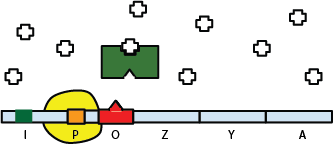
L'ensemble des gènes prenant part à la métabolisation du lactose et à son contrôle se trouvent dans la même zone physique. Elle se compose d'un gène répresseur I (pour Inhibitor) qui est toujours exprimé, de trois gènes de structure, lacZ, lacY et lacA et d'une région régulatrice contenant une zone promoteur P et une zone Opérateur O. La partie contenant le promoteur P, l'opérateur O et les gènes de structures est appelée un opéron, et dans ce cas précis, l'opéron lac.
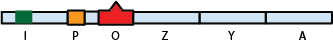
Structure de l'opéron lac
lacZ permet la synthèse de la béta-galactosidase qui convertit le lactose en deux monosaccharides, le glucose et le galactose; lacY la synthèse de la perméase qui facilite l'entrée du lactose dans la cellule, et lacA la transacétylase qui semble impliquée dans la suppression de certains résidus toxiques de la dégradation du lactose.
Le gène I permet la synthèse d'une protéine, le répresseur lac (représenté en vert sur le schéma ci-dessous). Cette protéine se lie à l'ADN au niveau de l'opérateur O. Au moment de la transcription des gènes lacZ, lacY et lacA, la présence du répresseur lac bloque la transcription ADN-ARN en empêchant la fixation sur l'ADN du complexe (en jaune) qui assure cette transcription.
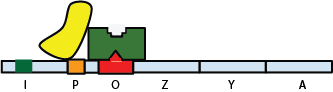
Fixation du répresseur lac sur l'opérateur
Mais en présence de lactose, celui-ci se fixe sur le répresseur lac changeant sa structure allostérique, et le rendant ainsi incapable de se fixer sur l'opérateur O. La transcription de l'ADN devient alors possible, et les gènes lacZ, lacY et lacA sont exprimés.
Inhibition du répresseur par le glucose
par modification allostérique
Il s'agit bien d'un contrôle négatif puisque les gènes sont exprimés sauf si le répresseur lac est présent. Il faut savoir qu'il peut exister des opérons, comme l'opéron arabinase qui peuvent exercer simultanément un contrôle positif ou négatif.
La régulation des protéines est un autre effet classique de l'expression, ou de la non-expression, d'un génotype en fonction de l'environnement. Le cas le plus visuel se rencontre chez le lapin de l'Himalaya ou chez le chat siamois. Chez ces animaux, la protéine (la tyrosinase) qui permet de colorer (en noir) les poils de l'animal est muté, et ne s'exprime qu'en dessous d'une certaine température. C'est ainsi que les parties froides du corps (les extrémités) de l'animal sont noires, alors que le reste du corps est blanc. A la naissance, l'ensemble du corps est blanc, puisque dans l'utérus la température est constante et élevée partout.


Les génomes de deux êtres humains pris au hasard sont semblables à 99.9%, c'est à dire que l'on observe sur 2x3 milliards de paires de bases une variation d'environ 1 pour 1000 soit seulement 2x3 millions de paires de base (ces valeurs sont à prendre avec précaution, seul l'ordre de grandeur est sûr, il est possible que le nombre de SNP soit en fait plus important). Sur ces variations, 90% d'entre elles consistent en une différence isolée d'un seul nucléotide à l'intérieur d'une séquence. C'était par exemple le cas pour l'anémie falciforme où une adénine est remplacée par une thymine à l'intérieur du gène HBB. Ces modifications isolées d'un seul nucléotide sont appelées SNP, de l'anglais Single Nucleotid Polymorphism. Pour qu'une variation dans le génome soit considéré comme étant un SNP, il faut que cette variation apparaisse dans au moins 1% de la population.
Exemple de variation SNP dans le gène HBB
On connaît aujourd'hui un grand nombre de SNPs situés dans les gènes (ou dans les zones des précurseurs des gènes) qui sont responsables de maladies génétiques. On a pour ces SNPs un vrai rapport de causalité, puisque l'on sait que c'est la modification du nucléotide qui entraîne directement la modification de la protéine et, par là même, la maladie.
Mais seulement 3 à 5% de l'ADN contient les gènes, un grand nombre de SNPs se trouvent en dehors des séquences codantes. Ces gènes ont aussi un intérêt car ils servent à établir des cartes génétique par associations pour identifier la position des gènes responsables des traits d'origine génétique. Il s'agit là d'une idée très ancienne et relativement simple. Lors de l'analyse d'un trait génétique, si l'on parvient à trouver une association statistique entre ce trait et un SNP dans une même lignée familiale, on a une très forte probabilité de trouver le gène responsable à proximité de ce SNP, en raison de la localité de la recombinaison lors de la méiose: le SNP "voyage avec" le gène, et la probabilité de rester co-localisé est une fonction de la distance entre les deux (c'est un sujet que nous avons déjà vu lors du paragraphe sur l'hypothèse des caractères indépendants).
Lien entre un SNP et un gène proche
De la même façon, cette technique a été largement utilisée pour faire des analyses par corrélation permettant à l'intérieur d'une même lignée familiale de prévoir, par exemple en diagnostic prénatal, la présence d'un allèle muté. Nous allons nous arrêter brièvement sur un exemple pour bien comprendre la différence entre les SNPs causaux et les SNPs qui sont seulement corrélés.
Nous allons reprendre comme exemple le cas de l'hémophilie A, une maladie non-autosome récessive transmise par le chromosome X. Dans le schéma suivant les individus de sexe masculin sont représentés par des carrés, ceux de sexe féminin par des ronds. Les figures en blanc sont des phénotypes sains, les figures en noir plein des phénotypes hémophiliques, les figures contenant un point noir sont des individus hétérozygotes phénotypiquement sains. On remarque évidemment que les hommes ne peuvent pas être hétérozygotes puisque, n'ayant qu'un chromosome X, ils sont soit atteints, soit sains. Les femmes en revanche peuvent être porteurs sains (rond blanc avec un point noir).
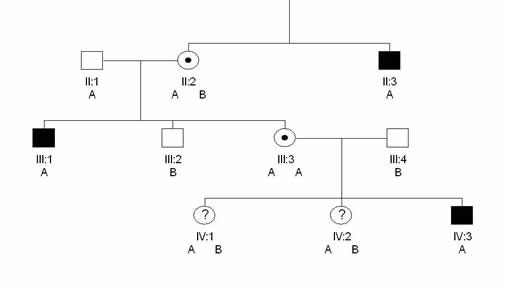
Pédigré et marqueur SNP
Extrait de practical-haemostasis.com
On suppose que dans une lignée familiale l'on a réussi à corréler avec
la maladie un SNP qui peut
avoir deux formes alléliques, A ou B. En génération II les deux
enfants sont tous deux porteurs de la maladie, l'individu II:3 étant
malade et l'individu II:2 porteur sain hétérozygote. On constate que
pour cette lignée familiale, l'hémophilie est associée à l'allèle A du
SNP. Ainsi II:3 (malade) possède l'allèle A du SNP et II:2 possède
l'allèle A du SNP pour un chromosome X et l'allèle B pour l'autre, tout
cela étant cohérent avec le fait qu'elle ne soit que porteur sain.
Supposons maintenant que II:2 ait des enfants avec II:1,
individu mâle non malade mais néanmoins porteur d'un SNP A (ce qui est
parfaitement possible, le SNP A n'étant corrélé avec
l'hémophilie que dans l'autre branche familiale). En génération III,
on sait de façon certaine que les individus mâles
porteurs d'un SNP B seront obligatoirement sains (car il s'agit
forcément du B associé au "gène sain" de la mère, à condition bien sûr
que la recombinaison n'ait pas "cassé" le chromosome entre le marqueur
et le gène, mais cela est extrêmement improbable s'ils sont proches),
que les mâles
SNP A seront malades (car il s'agira là aussi du SNP-A de la mère
associé au gène muté) que les
femmes SNP A-A seront forcément porteurs sains de la maladie et les
femmes SNP A-B seront homozygotes saines. En revanche, on constate
que sur l'individu III:3, il est devenu impossible de distinguer le
gène sain du gène muté puisque les deux sont marqués par un SNP
A. Ainsi, en génération IV, il serait impossible de faire un
diagnostic prénatal sur IV:3 qui peut aussi bien être malade que sain,
avec dans les deux cas un SNP A, pas plus que l'on ne peut savoir si
IV:1 et IV:2 sont homozygotes sains ou hétérozygotes.
On voit donc bien que les SNP sont des marqueurs utiles, mais limités car ils possèdent peu de formes alléliques. Une solution consiste à en associer plusieurs qui seraient tous proches du gène responsable (si l'on utilise n SNP, on a 2n configurations alléliques possibles ), mais cela signifie qu'il faut disposer d'une très forte densité de SNP si l'on veut garder une grande fiabilité dans l'analyse.
Rappelons nous bien une chose: si un SNP intra-génique établit (en général) une relation de cause à effet absolue (tout individu porteur de l'allèle mutée aura un gène qui produira une protéine déficiente), un SNP extra-génique ne permet que d'établir des relations de corrélation au sein d'une même lignée. Nous verrons plus loin dans le cadre des hypothèses des études GWAS comment on peut essayer malgré tout de lier des SNPs avec des caractéristiques phénotypiques dans des études plus globales sur des populations de grande taille.
Les SNP sont devenus des éléments essentiels pour l'identification génotypique. Les méthodes industrielles de type MIP (Molecular Inversion Probes) d'échantillonage génétique sont en passe de devenir les méthodes dominantes et permettent de traiter de grandes quantités de loci à une vitesse extrêmement élevée et pour des coûts très faibles. On parle actuellement de quelques centaines d'euros, et probablement un jour quelques dizaines d'euros pour analyser jusqu'à 10000000 de SNP dans le génome humain, comme le fait par exemple le chip d'Affymetrix. Dans le cas du maïs les chips d'Illumina permettent d'analyser aujourd'hui 50000 SNP, mais ce nombre va augmenter très rapidement.

Chip SNP6.0 d'Affymetrix
Les premières méthodes utilisées pour détecter les SNP consistaient à construire une séquence exactement complémentaire de la zone d'ADN à tester et à regarder si la ligature entre la sonde et l'ADN se faisait.
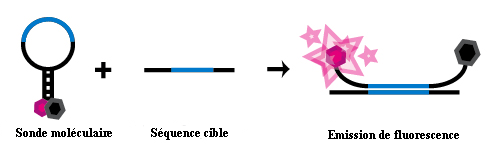
Méthode élémentaire de détection de SNP
Lorsque la séquence de la sonde était exactement complémentaire de la séquence de l'ADN, la liaison était totale et dépliait la boucle, libérant le marqueur fluorescent. Ces techniques ne permettaient de tester qu'un seul allèle à la fois. Les méthodes MIP, plus complexes, permettent de tester simultanément plusieurs allèles, et ce sur de nombreux sites simultanément
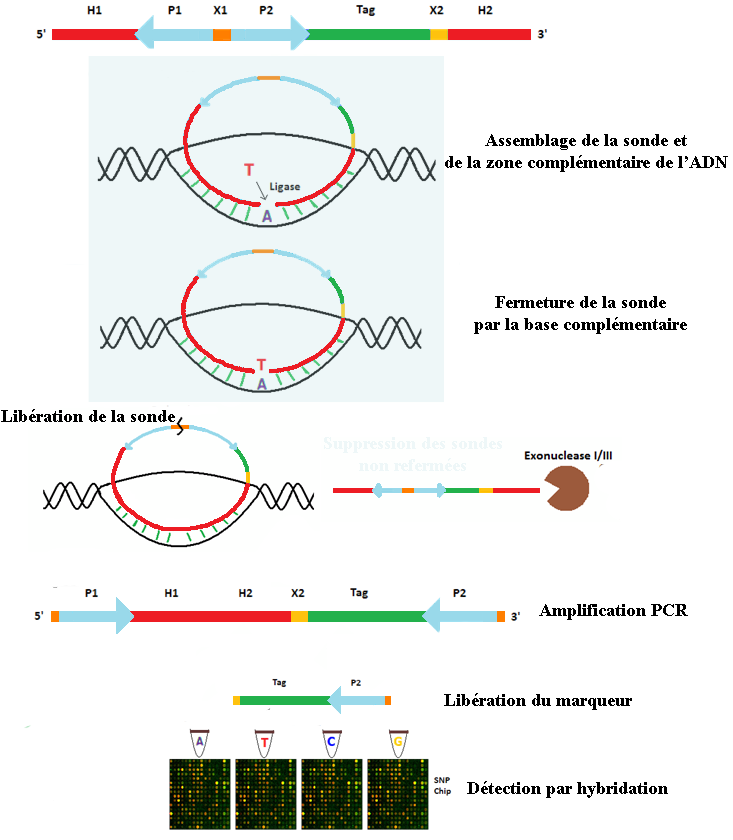
Principe de la méthode MIP
Il faut en conclusion bien se souvenir de la réalité physique des analyses SNP: il s'agit au final d'une analyse du niveau de fluorescence lié à la libération d'une sonde situé dans un brin complémentaire d'ADN: mêmes si les méthodes sont de plus en plus fiables, il existe de nombreuses possibilités d'erreurs ou d'imprécisions (décalage des nucléotides, imprécision dans la valeur du niveau de fluorescence, etc.)
La variabilité du nombre de copies (CNV en anglais) est une forme de variation structurelle du génome qui vient de la multiplication d'une zone relativement longue (d'un millier à plusieurs millions de bases) du génome, ou, dans des cas moins fréquents la disparition d'un certain nombre de copies.
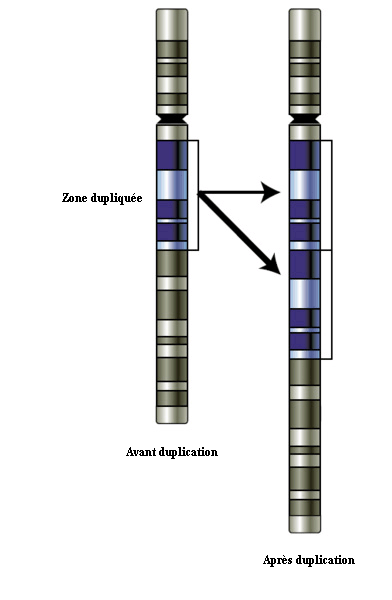
Duplication d'une zone du génome
Les CNV peuvent être héritées, ou être dues à des mutations "de novo", et, comme les SNP, elles peuvent se produire au niveau de l'ADN codant ou de l'ADN non codant. Au niveau de l'ADN codant, un des exemples marquants est la réplication de gènes entiers. Ainsi le gène codant l'amylase salivaire chez l'homme existe suivant les individus en 6 à 15 copies, alors qu'il n'en existe que 2 exemplaires chez le chimpanzé. Il s'agit probablement d'une évolution liée à une alimentation plus riche en amidon chez l'homme.
Les CNV ont été relativement peu étudiées jusqu'ici. On pense maintenant qu'ils ont une importance qui peut être grande en tant que marqueurs génétiques, mais aussi dans l'origine de certaines maladies génétiques. On pense par exemple que la réduction du nombre de copies du gène DEFB4 qui code la protéine SAP1 (skin antimicrobial peptide 1) est un facteur de prédisposition génétique à la maladie de Crohn. Alors que le nombre normal de copies de ce gène est 4, 70% des malades souffrant de la maladie de Crohn semblent avoir 3 (ou moins) copies de ce gène (Jager 2011). Les CNV sont extrêmement fréquents dans le génome humain et on a même découvert des cas de CNV entre de vrais jumeaux.
Certains fabricants de chip SNP ont maintenant intégré à côté de la détection de SNP la détection de CNV. C'est par exemple le cas de l'Affymetrix 6 qui associe à la détection d'un million de SNP un million de sondes permettant de réaliser des détections de CNV.
Les CNV ne sont pas les seules variations structurelles du génome. Il en existe bien d'autres (voir ci-dessous) qui semblent toutes avoir une importance. L'étude des variations structurelles est un des champs en cours d'explorations dans le cadre de la compréhension du génome.
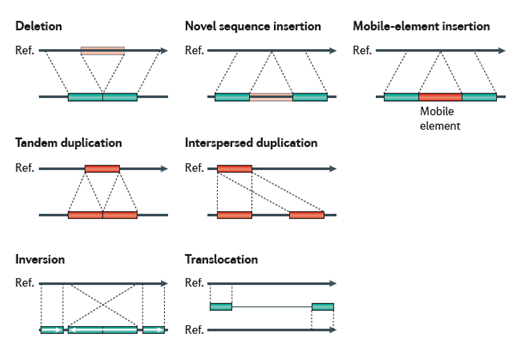
Différentes modifications structurelles du génome
(extrait de Jager 2011)
Les microsatellites (appelés aussi SSR: Simple Sequence Repeat ou STR: Short Tandem Repeat) sont des répétitions de séquences de 2 à 6 paires de bases dans l'ADN. Les séquences répétés sont généralement simples et ne dépassent généralement pas 4 nucléotides. Un des microsatellites les plus fréquents dans le génome humain est la répétition du dinucléotides CA, qui se rencontre environ tous les 2000 ou 3000 paires de bases. Le nombre de répétitions de ces séquences varie en général de 3 à 100. Pour un locus donné, il existe en général de très nombreux allèles, ce qui permet une identification génétique de filiation extrêmement fiable et en fait d'excellents marqueurs génétiques. Pour cette raison, les microsatellites sont utilisés dans le cadre des analyses de police scientifique (la célèbre base d'ADN du FBI, le CODIS, est basée sur l'analyse des STR de treize loci).
Comme pour les SNP et les CNV, les STR peuvent se produire aussi bien dans des gènes, que dans des zones promotrices ou non codantes. On pense que dans les mammifères 20 à 40% des gènes contiennent des séquences répétées de tri-nucléotides qui augmentent la proportion d'un des acides aminés dans la protéine codée. Ceci entraîne évidemment des modifications phénotypiques. Un des exemples classiques est la répétition de certaines séquences au sein du gène RUNX2 (codant une protéine qui est facteur de transcription associé à la différentiation des ostéoblastes, les cellules osseuses germinales) chez le chien. Ces répétitions entraînent des modifications dans la longueur du museau. Chez l'homme, le nombre de répétitions de la polyalanine dans le gène HoxA13 entraîne des malformations au niveau du développement physique (mains/pieds et appareil urétro-génital). Pour des raisons moins claires, la présence de microsatellites dans les introns des gènes semble avoir aussi une grande influence, puisque l'on sait par exemple que l'ataxie de Friedreich est causée par une répétition trop importante d'un codon GAA dans un intron du gène FXN codant la frataxine.
Nous allons voir sur le même exemple que ci-dessus pourquoi les microsatellites sont de meilleurs marqueurs génétiques que les SNP pour faire des analyses de corrélation. Reprenons le même schéma, mais ici le gène muté est associé à 17 répétitions d'un STR.
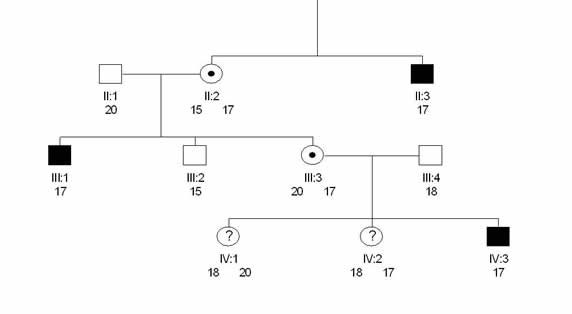
Pédigré et marqueur STR
En génération II, l'individu II:3 est bien porteur d'un gène muté, repéré par les 17 répétitions du STR, l'individu II:2 est hétérozygote porteur sain avec un gène portant le marqueur 17 et un autre gène portant un marqueur 15. En épousant un individu portant un marqueur 20, nous avons en génération III un individu mâle (malade) portant le marqueur 17, un mâle sain portant le marqueur 15, et une femme porteur sain avec un marqueur 17 et un marqueur 20. Lorsque ce porteur sain épouse un individu sain portant un marqueur 18, nous sommes maintenant capables e savoir qu'en génération IV, un mâle porteur du marqueur 17 sera malade (IV:3), que l'individu IV:2 est hétérozygote porteur sain et que IV:1 est totalement sain (pas de marqueur 17). On peut ainsi lever totalement les ambiguïtés qui existaient avec les SNP, car les marqueurs STR possèdent beaucoup plus d'allèles, et il est donc moins probable de rencontrer deux individus portant le même allèle associé à des phénotypes différents.
Il est évidemment possible de considérer l'humanité dans son ensemble comme une seule grande "famille" dont les origines uniques seraient très anciennes. Certaines analyses faites pour des populations ayant eu peu de diversité génétique (comme la population finlandaise par exemple), ont montré la validité de cette hypothèse; cependant elle nécessite pour être efficace une densité exceptionnelle de SNP: un SNP toutes les 5000 bases (soit 500000 SNPs sur le génome humain) ne donne qu'un corrélation r2 de 0.1, bien trop faible. Ce type d'analyse est appelé Genome Wide Association Studies (GWAS) et en est encore à ses débuts (2006).
Pour réaliser ce type d'analyse, on utilise deux échantillons, une population de test qui est composée d'individus présentant le trait phénotypique que l'on cherche à corréler avec des SNP, et une population de contrôle qui est supposée statistiquement uniforme. On mesure alors pour tous les SNPs les répartitions des allèles et on cherche les SNPs pour lesquels les ratios entre la population de test et la population de contrôle sont les plus éloignés de 1. On calcule ensuite pour chacun des SNP la p-value (voir ci-dessous) associée et l'on représente la graphe de Manhattan (Manhattan plot) donnant -log(p_value) en fonction du SNP.
Voyons cela sur un exemple concret. On dispose d'une population test de 4000 individus et d'une population de contrôle de 6000 individus sur chacune de ces populations on teste deux SNP, qui sont di-alléliques.
| SNP1 | SNP2 | |
| Ptest, nombre allèle 1 | 2104 | 1648 |
| Ptest, fréquence allèle 1 | 2104/4000=52.6% | 1648/4000=41.2% |
| Pcontrole, nombre allèle 1 | 2676 | 2532 |
| Pcontrole, fréquence allèle 1 | 2676/6000=44.6% | 2532/6000=42.2% |
| Ratio | 52.6/44.6=1.18 | 41.2/42.2=0.98 |
| p-value | 5.10-15 | 0.33 |
On voit que dans ce cas (extrait d'une étude réelle), il y a de très fortes chances que le SNP1 soit corrélé avec le trait phénotypique recherché, alors que le SNP2 a très peu de chance d'être significatif.
La p-value, traduit parfois en français par niveau de signification (Saporta), est une mesure de la confiance avec laquelle on peut rejeter l'hypothèse H0, (ou null hypothesis en anglais) dans un test statistique. Considérons un exemple simple: supposons que nous voulions tester si une pièce est biaisée ou non. Supposons que sur 20 lancers, nous obtenions 14 fois "face". L'hypothèse H0 que nous voulons accepter ou rejeter est la pièce n'est pas biaisée. La p-value de ce tirage est la probabilité d'obtenir 14 fois "face" ou plus sur 20 lancers avec une pièce bien équilibrée. Dans ce cas nous obtenons 60460/1048576=0.058, soit environ 6%. Il s'agit de la probabilité qu'une pièce non biaisée nous donne un tirage contenant 14 fois "face" ou plus. On choisit ensuite un niveau de risque, et l'on rejette l'hypothèse H0 si la p-value est inférieure à ce niveau de risque. Si par exemple j'avais choisi 10% de niveau de risque, je pourrais rejeter dans ce cas H0 (avec un risque de 10%). En revanche si mon niveau de risque est de 5%, je ne peux pas rejeter H0 (ce qui ne signifie en aucun cas que H0 est correct mais simplement que je n'ai pas assez de données pour conclure).Il faut donc retenir que plus la p-value est petite, et plus notre risque est faible de rejeter H0. Dans le cas des SNP, l'hypothèse H0 est "le SNP n'est pas corrélé avec le trait phénotypique", et plus la valeur de -log(p_value) est grande, plus forte est donc notre confiance dans le fait que le SNP et le trait phénotypique sont corrélés.
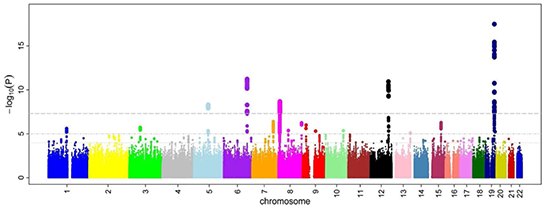
Représentation Manhattan
En abscisse, la position du SNP dans le génome
En ordonné, l'opposé du log de la p-value
L'utilisation des SNP (ou d'autres marqueurs) à très haute densité dans les GWAS a permis de réaliser de sérieuses avancées dans le domaine de l'analyse des origines génétiques de certaines maladies. Elle a en particulier permis de montrer que la majorité des traits transmissibles génétiquement fonctionnait sur un modèle polygénique, c'est à dire qu'un grand nombre de gènes sont généralement impliqués dans l'expression phénotypique.
Le fonctionnement polygénique de certains caractères héritables est connu depuis 1909, et est lié au caractère additif de certains gènes dans l'expression d'un phénotype. L'expérience fondatrice de Nilson-Ehle avait par exemple montré que le croisement de blé blanc et de blé rouge permettait d'obtenir plusieurs nuances intermédiaires entre le blanc et le rouge qui ne pouvait s'expliquer que dans la mesure où plusieurs gènes étaient impliqués, et où leurs contributions étaient additives. On connaît aujourd'hui plusieurs prédispositions génétiques (comme celle au diabète tardif) qui s'explique par des modèles polygéniques.
Les modèles polygéniques expliquent aussi l'intérêt des méthodes GWAS qui recherchent des corrélations sur de très grands nombres de SNP sans se préoccuper de trouver un gène candidat responsable.
Il faut noter qu'une étude récente (An Open Access Database of Genome-wide Association Results) semble montrer que les SNPs significatifs connus au jour de l'étude se trouvaient pour 40% d'entre eux à l'intérieur des gènes, et que seuls 32% de SNPs à plus de 60kb d'un gène de la base RefSeq semblaient avoir une influence sur les traits phénotypiques recherchés.
D'autre part, on a également pu montrer la validité de l'hypothèse CD-CV (Common disease, common variation). Cette hypothèse suggérait que la plupart des maladie génétiques "communes" sont liés à des mutations "communes", c'est à dire des mutations relativement fréquentes à l'intérieur du génome. Depuis plusieurs années ces méthodes ont permis de valider la prédisposition génétique pour des maladies aussi variées que la maladie d'Alzheimer, le diabète ou les désordres bipolaires.
Il faut rester très prudent dans l'utilisation des études de type GWAS. Tout récemment un article publié dans Science ( Genetic signatures of exceptional longevity in humans, 01/07/2010 ) a été très critiqué par la communauté, et finalement retiré par les auteurs (12/11/2010), avant d'être corrigé et re-publié dans PLOS ONE le 18/01/2012.
Les analyses GWAS ont encore aujourd'hui des détracteurs pour de nombreuses raisons, qui tiennent à la fois au fond et à la forme.
Un des problèmes classiques des analyses GWAS et bien connu en statistiques est le problème des comparaisons multiples. Supposons par exemple que l'on souhaite vérifier si une pièce est truquée ou bien équilibrée. Pour cela on la lance 10 fois, et si on constate par exemple qu'elle tombe 9 fois sur "face", on estimera qu'elle est biaisée, et effectivement la probabilité de 9 "face" sur 10 tirages n'est que de 1%. Mais supposons que l'on décide de tester un lot de 100 pièces par cette méthode: même si les 100 pièces sont bien équilibrées, nous avons environ 66% de chance d'en trouver une que nous déclarerons biaisée dans le lot. Les méthodes "naïves" qui s'appliquent pour une pièce ne s'applique pas pour un lot de 100 pièces. La probabilité de trouver de faux-positifs "naïfs" dans les associations SNP/phénotype est d'autant plus importante que l'on teste beaucoup de SNPs...
Le téléchargement ou la reproduction des documents et photographies
présents sur ce site sont autorisés à condition que leur origine soit
explicitement mentionnée et que leur utilisation
se limite à des fins non commerciales, notamment de recherche,
d'éducation et d'enseignement.
Tous droits réservés.
Dernière modification: 09:48, 22/02/2024